
Code
cd ~/work/PROJECTS/SCPWI/RAW_DATA
wget https://galaxy.migale.inrae.fr/api/datasets/730f755068bc87c9/display?to_ext=tar -O IC_Auz.tar
wget https://galaxy.migale.inrae.fr/api/datasets/8918474ce4f0a666/display?to_ext=tar -O IC_Bre.tarThis document is a report of the analyses performed. You will find all the code used to analyze these data. The version of the tools (maybe in code chunks) and their references are indicated, for questions of reproducibility.
The aim of this project is to obtain to amount of reads mapped on each reference present in the samples.
Deliverables agreed at the preliminary meeting (Table 1).
| Definition | Status | |
|---|---|---|
| 1 | HTML report | ✔️ |
All data is managed by the migale facility for the duration of the project. Once the project is over, the Migale facility does not keep your data. We will provide you with the raw data and associated metadata that will be deposited on public repositories before the results are used. We can guide you in the submission process. We will then decide which files to keep, knowing that this report will also be provided to you and that the analyses can be replayed if needed.
The raw data were stored in the abaca server and on a dedicated space on the front server.
cd ~/work/PROJECTS/SCPWI/RAW_DATA
wget https://galaxy.migale.inrae.fr/api/datasets/730f755068bc87c9/display?to_ext=tar -O IC_Auz.tar
wget https://galaxy.migale.inrae.fr/api/datasets/8918474ce4f0a666/display?to_ext=tar -O IC_Bre.tarThe files need to be renamed and compressed.
cd ~/work/PROJECTS/SCPWI/RAW_DATA
tar xvf IC_Bre.tar
mkdir IC_Bre IC_Auz
for i in *.fastq ; do id=$(basename $i |cut -d '_' -f 2) ;\cp $i IC_Bre/${id}.fastq ; done
cd IC_Bre/
pigz *.fastq
cd ../
rm -f *.fastq
tar xvf IC_Auz.tar
for i in *.fastq ; do id=$(basename $i |cut -d '_' -f 2) ;\cp $i IC_Auz/${id}.fastq ; done
cd IC_Auz
pigz *.fastqAuzeville and Breteniere.conda activate seqkit-2.0.0
seqkit stat *.fasta
file format type num_seqs sum_len min_len avg_len max_len
SynCom_Ref16S_Auzeville.fasta FASTA DNA 14 13,500 920 964.3 1,011
SynCom_Ref16S_Breteniere.fasta FASTA DNA 11 10,672 799 970.2 1,070
conda deactivaterefs <- read.csv2(file = "data/GTDB_16S_release220_202411.taxonomy", sep = "\t", header = TRUE, col.names = c("GTDB_ID", "Taxonomy")) %>% mutate(GTDB_ID = str_trim(GTDB_ID), Taxonomy = str_trim(Taxonomy))We check if reference sequences are present in the GTDB databank by blasting them.
conda activate blast-2.15.0
makeblastdb -in GTDB_16S_release220_202411/GTDB_16S_release220_202411.fasta -dbtype nucl
blastn -query SynCom_Ref16S_Auzeville.fasta -db GTDB_16S_release220_202411/GTDB_16S_release220_202411.fasta -outfmt "6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore stitle" -out SynCom_Ref16S_Auzeville_vs_GTDB.blast -max_target_seqs 1
blastn -query SynCom_Ref16S_Breteniere.fasta -db GTDB_16S_release220_202411/GTDB_16S_release220_202411.fasta -outfmt "6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore stitle" -out SynCom_Ref16S_Breteniere_vs_GTDB.blast -max_target_seqs 1
conda deactivated <- read.csv2(file = "data/SynCom_Ref16S_Auzeville_vs_GTDB.blast", sep = "\t", header = FALSE, col.names = c("qseqid", "sseqid", "pident", "length", "mismatch", "gapopen", "qstart", "qend", "sstart", "send", "evalue", "bitscore", "stitle" ))
d[,-ncol(d)] %>% reactable::reactable(rownames = FALSE)d <- read.csv2(file = "data/SynCom_Ref16S_Breteniere_vs_GTDB.blast", sep = "\t", header = FALSE, col.names = c("qseqid", "sseqid", "pident", "length", "mismatch", "gapopen", "qstart", "qend", "sstart", "send", "evalue", "bitscore", "stitle" ))
d[,-ncol(d)] %>% reactable::reactable(rownames = FALSE)The hits have a very high percent of identity and an expected length, suggesting the references are comprised in the GTDB databank. We can use it for taxonomic affiliation without losing information.
conda activate seqkit-2.0.0
cd /home/orue/work/PROJECTS/SCPWI/RAW_DATA/IC_Auz
seqkit stat --all -T -j 24 *.gz >> seqkit.txt
conda deactivate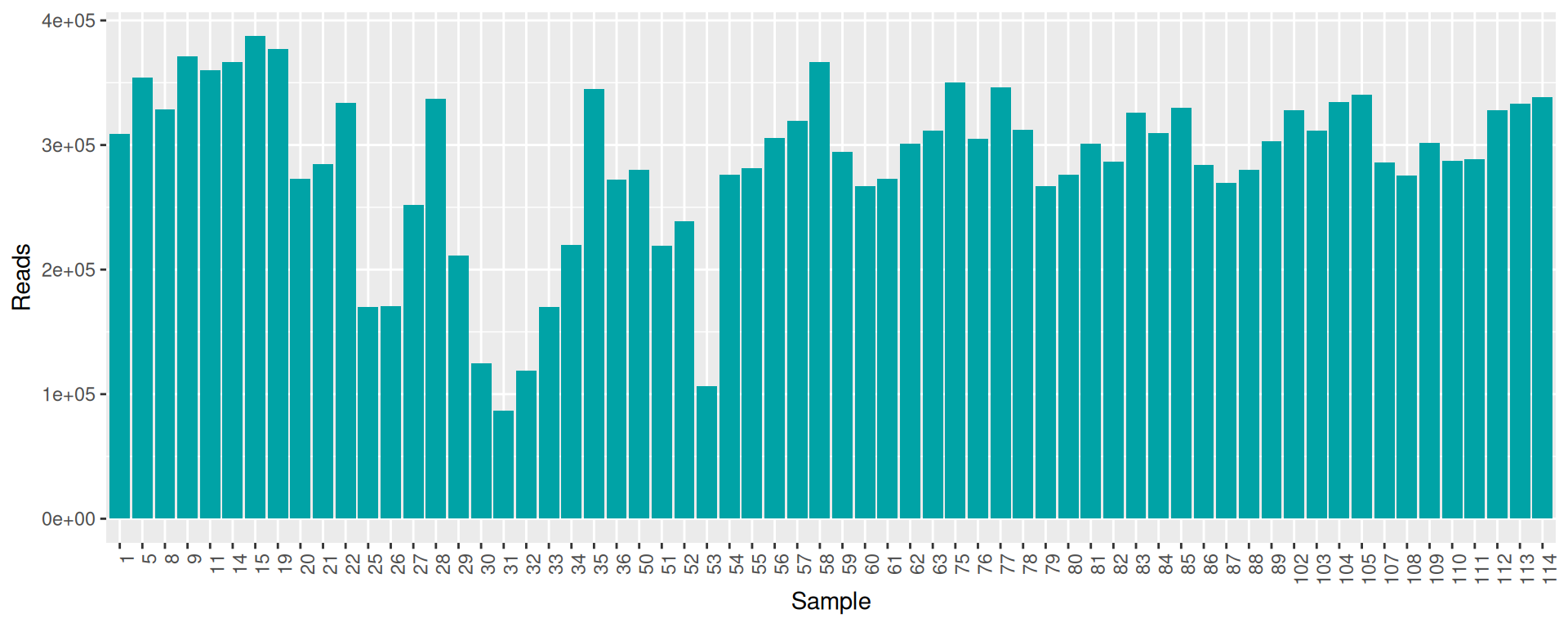
raw_data %>% select(sample, num_seqs, sum_len, min_len, avg_len, max_len) %>% arrange(as.integer(sample)) %>% datatable()conda activate seqkit-2.0.0
cd /home/orue/work/PROJECTS/SCPWI/RAW_DATA/IC_Bre
seqkit stat --all -T -j 24 *.gz >> seqkit.txt
conda deactivate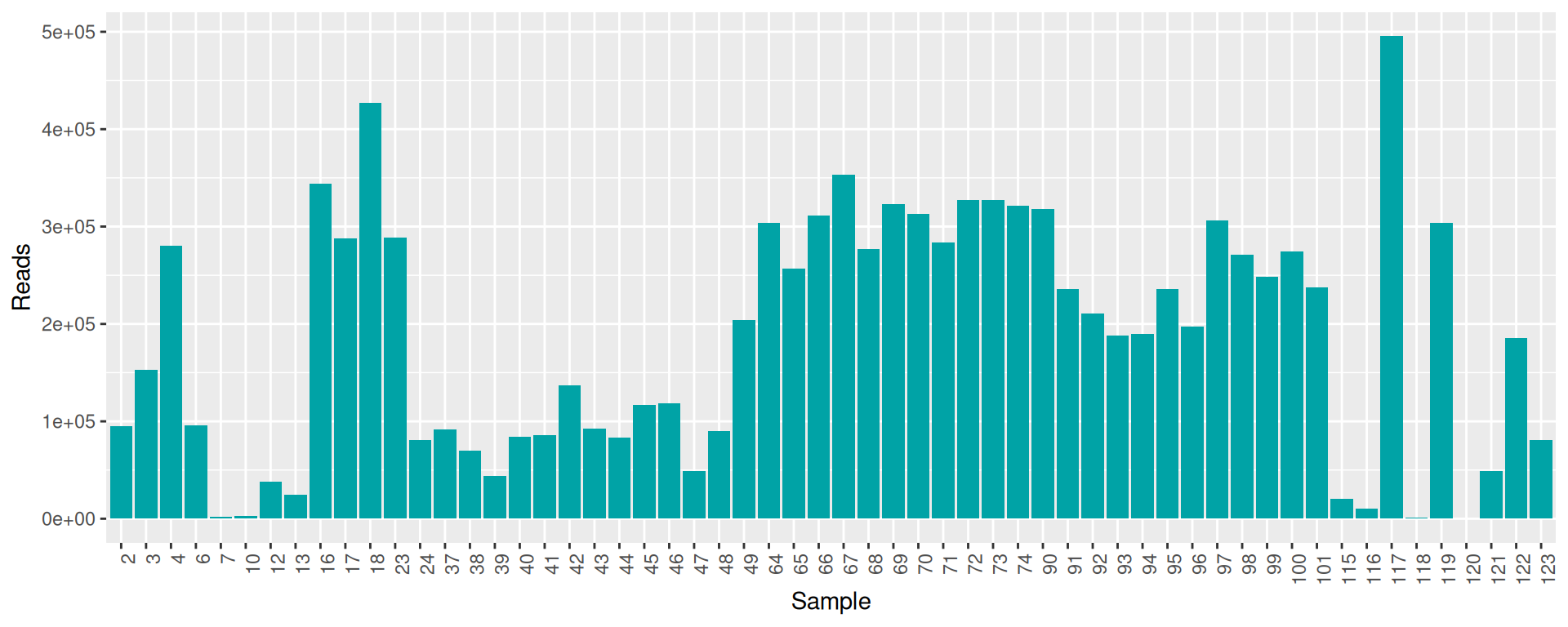
raw_data %>% select(sample, num_seqs, sum_len, min_len, avg_len, max_len) %>% arrange(as.integer(sample)) %>% datatable(rownames = F)Some samples have a low number of reads. We keep them, but we could remove them at this step.
for i in RAW_DATA/IC_Auz/*.fastq.gz ; do echo "conda activate fastqc-0.11.9 && fastqc -t 8 $i -o QC/Auzeville/FASTQC && conda deactivate" >> fastqc_auz.sh ; done
qarray -cwd -V -N fastqc -pe thread 8 -o LOGS -e LOGS fastqc_auz.sh
qsub -cwd -V -N multiqc -o LOGS -e LOGS -b y "conda activate multiqc-1.11 && multiqc QC/Auzeville/FASTQC -o QC/Auzeville/MULTIQC && conda deactivate"Here is the MultiQC report
for i in RAW_DATA/IC_Bre/*.fastq.gz ; do echo "conda activate fastqc-0.11.9 && fastqc -t 8 $i -o QC/Breteniere/FASTQC && conda deactivate" >> fastqc_bre.sh ; done
qarray -cwd -V -N fastqc -pe thread 8 -o LOGS -e LOGS fastqc_bre.sh
qsub -cwd -V -N multiqc -o LOGS -e LOGS -b y "conda activate multiqc-1.11 && multiqc QC/Breteniere/FASTQC -o QC/Breteniere/MULTIQC && conda deactivate"Here is the MultiQC report
The metrics are expected for Nanopore reads. The quality is even quite good. The length distribution of reads shows short and very long reads that should not be present. We will remove them.
for s in RAW_DATA/IC_Auz/*.fastq.gz ; do id=$(echo $(basename $s |cut -d '.' -f 1)) ; NanoPlot --outdir QC/Auzeville/NANOPLOT/ --prefix ${id} --tsv_stats --info_in_report --format png --fastq $s --threads 16 ; done
for s in RAW_DATA/IC_Auz/*.fastq.gz ; do id=$(echo $(basename $s |cut -d '.' -f 1)) ; NanoStat --fastq $s --outdir QC/Auzeville/NANOSTATS/ -n ${id} -t 16 ; donefor s in RAW_DATA/IC_Bre/*.fastq.gz ; do id=$(echo $(basename $s |cut -d '.' -f 1)) ; NanoPlot --outdir QC/Breteniere/NANOPLOT/ --prefix ${id} --tsv_stats --info_in_report --format png --fastq $s --threads 16 ; done
for s in RAW_DATA/IC_Bre/*.fastq.gz ; do id=$(echo $(basename $s |cut -d '.' -f 1)) ; NanoStat --fastq $s --outdir QC/Breteniere/NANOSTATS/ -n ${id} -t 16 ; doneNanoplot and Nanostats give other quality metrics that I don’t show here because nothing abnormal was detected.
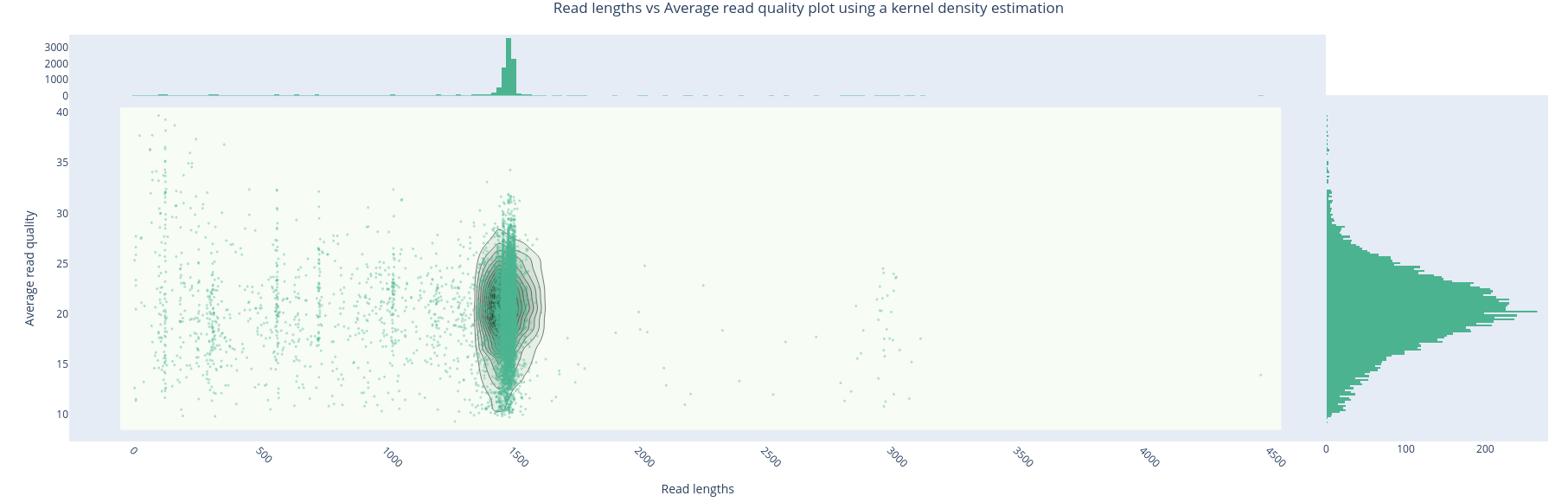
Filters used:
conda activate chopper-0.9.0
for s in RAW_DATA/IC_Auz/*.fastq.gz ; do id=$(echo $(basename $s |cut -d '.' -f 1)) ; gunzip -c $s | chopper -t 16 -q 10 --mingc 0.3 --maxgc 0.7 --minlength 800 --maxlength 2000 | gzip > CLEANING/Auzeville/${id}.fastq.gz ; done conda activate seqkit-2.0.0
cd /home/orue/work/PROJECTS/SCPWI/CLEANING/Auzeville
seqkit stat --all -T -j 24 *.gz >> seqkit.txt
conda deactivate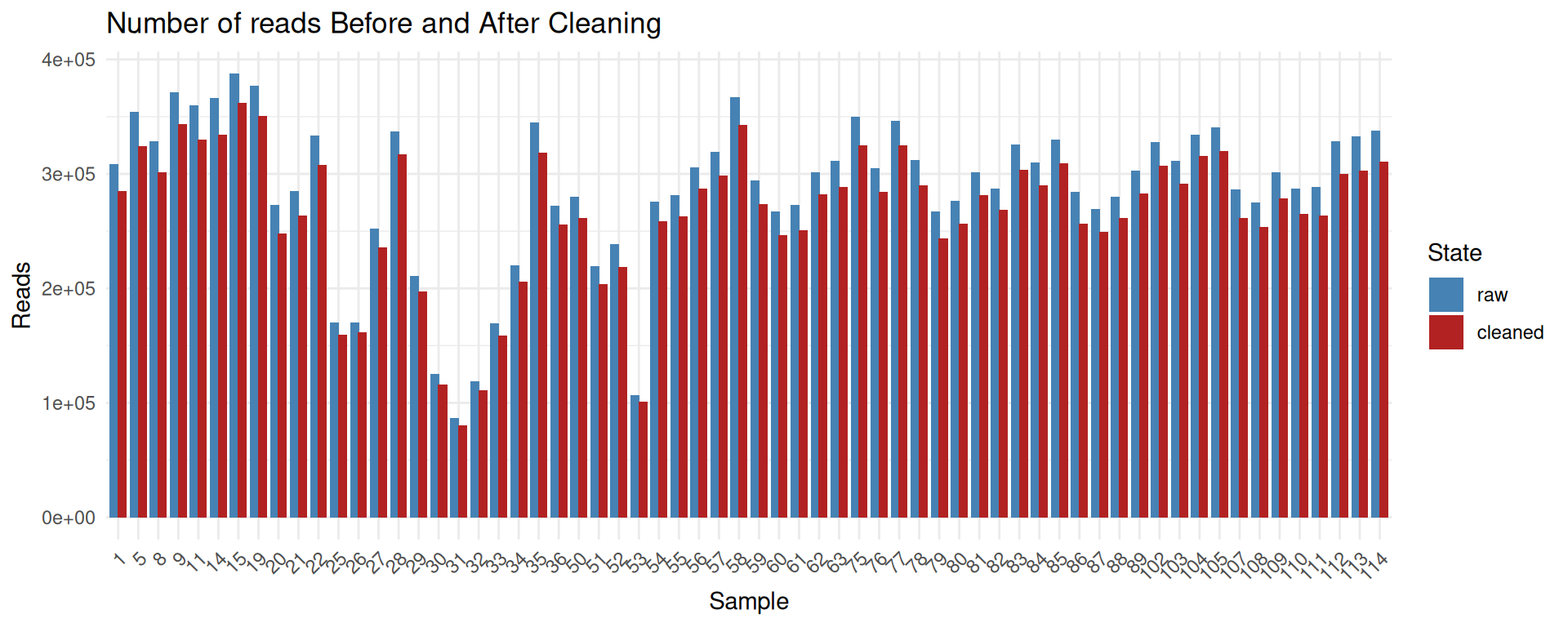
raw_data <- read.csv2("data/seqkit_Auz.txt", stringsAsFactors = F, sep="", strip.white=T, dec=".") %>% as_tibble() %>% mutate(sample = basename(gsub('.fastq.gz$', '', file))) %>% distinct(sample, .keep_all = TRUE) %>% select(-file) %>% mutate(sum_len = as.numeric(gsub(",", "", sum_len))) %>% arrange(as.integer(sample)) %>% mutate(state = "raw", sum_len = as.integer(sum_len)) %>% select(sum_len, sample, state)
cleaned_data <- read.csv2("data/seqkit_Auz_cleaned.txt", stringsAsFactors = F, sep="", strip.white=T, dec=".") %>% as_tibble() %>% mutate(sample = basename(gsub('.fastq.gz$', '', file))) %>% distinct(sample, .keep_all = TRUE) %>% select(-file) %>% mutate(num_seqs = as.numeric(gsub(",", "", num_seqs))) %>% arrange(as.integer(sample)) %>% mutate(state = "cleaned", sum_len = as.integer(sum_len)) %>% select(sum_len, sample, state)
combined_data <- bind_rows(raw_data, cleaned_data)
combined_data <- combined_data %>%
mutate(state = factor(state, levels = c("raw", "cleaned")))
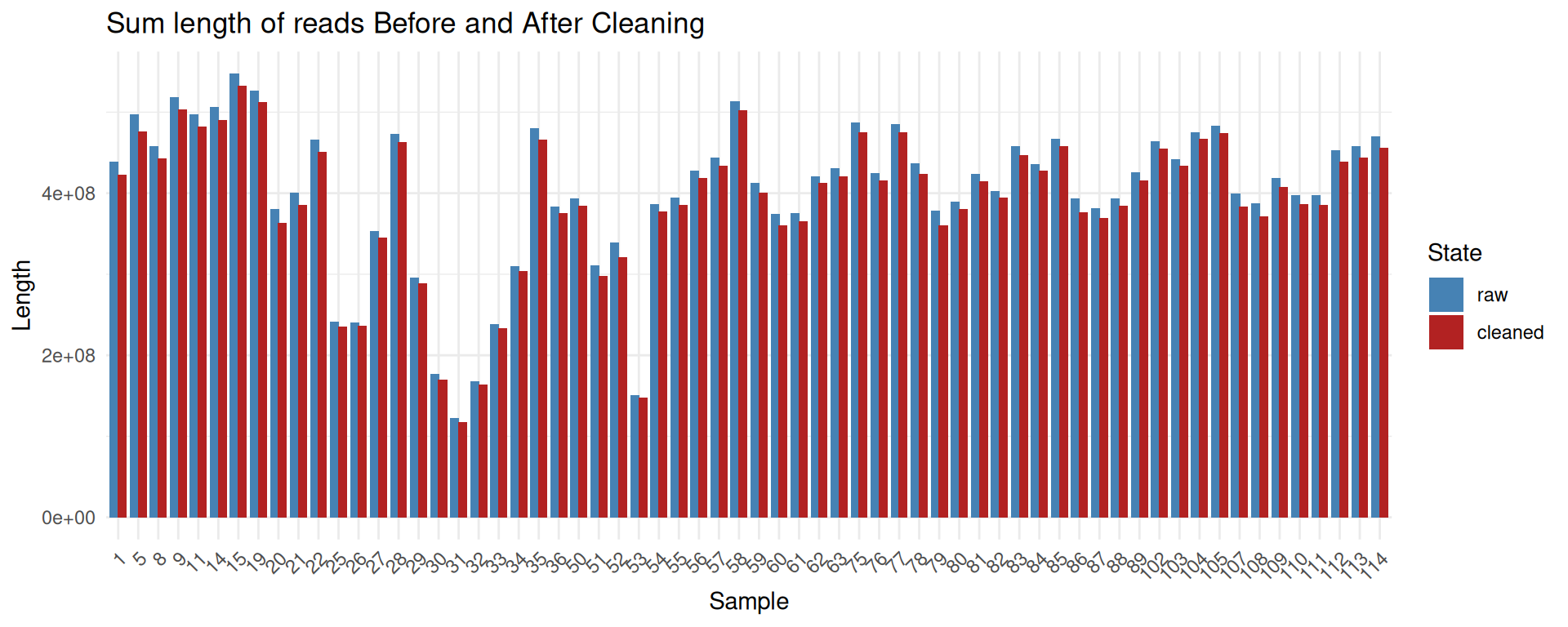
ggplot(combined_data, aes(x=reorder(sample, sort(as.numeric(sample))), y = sum_len, fill = state)) +
geom_bar(stat = "identity", position = position_dodge(width = 0.8)) +
scale_fill_manual(values = c("raw" = "steelblue", "cleaned" = "firebrick")) +
labs(
x = "Sample",
y = "Length",
fill = "State",
title = "Sum length of reads Before and After Cleaning"
) +
theme_minimal() +
theme(
axis.text.x = element_text(angle = 45, hjust = 1)
)
conda activate chopper-0.9.0
for s in RAW_DATA/IC_Bre/*.fastq.gz ; do id=$(echo $(basename $s |cut -d '.' -f 1)) ; gunzip -c $s | chopper -t 16 -q 10 --mingc 0.3 --maxgc 0.7 --minlength 800 --maxlength 2000 | gzip > CLEANING/Breteniere/${id}.fastq.gz ; done
conda deactivateconda activate seqkit-2.0.0
cd /home/orue/work/PROJECTS/SCPWI/CLEANING/Breteniere
seqkit stat --all -T -j 24 *.gz >> seqkit.txt
conda deactivate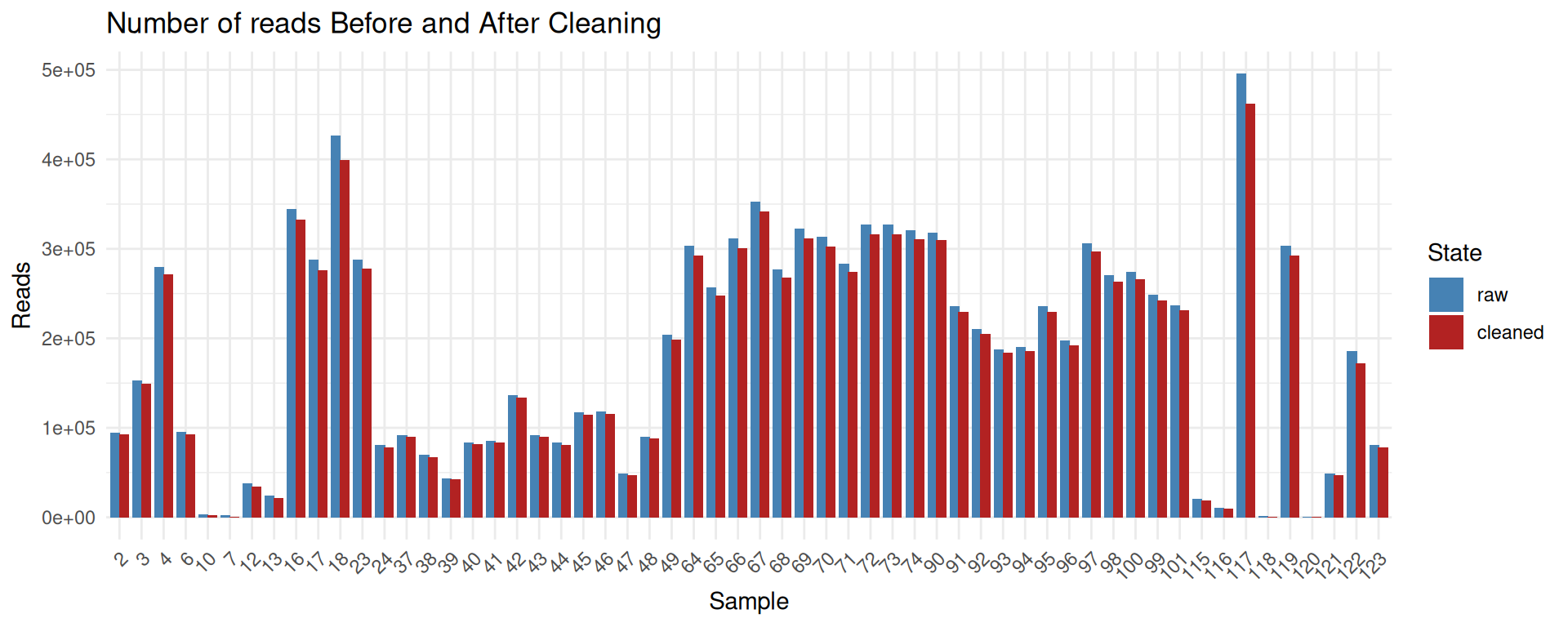
raw_data <- read.csv2("data/seqkit_Bre.txt", stringsAsFactors = F, sep="", strip.white=T, dec=".") %>% as_tibble() %>% mutate(sample = basename(gsub('.fastq.gz$', '', file))) %>% distinct(sample, .keep_all = TRUE) %>% select(-file) %>% mutate(sum_len = as.numeric(gsub(",", "", sum_len))) %>% arrange(as.integer(sample)) %>% mutate(state = "raw", sum_len = as.integer(sum_len)) %>% select(sum_len, sample, state)
cleaned_data <- read.csv2("data/seqkit_Bre_cleaned.txt", stringsAsFactors = F, sep="", strip.white=T, dec=".") %>% as_tibble() %>% mutate(sample = basename(gsub('.fastq.gz$', '', file))) %>% distinct(sample, .keep_all = TRUE) %>% select(-file) %>% mutate(num_seqs = as.numeric(gsub(",", "", num_seqs))) %>% arrange(as.integer(sample)) %>% mutate(state = "cleaned", sum_len = as.integer(sum_len)) %>% select(sum_len, sample, state)
combined_data <- bind_rows(raw_data, cleaned_data)
combined_data <- combined_data %>%
mutate(state = factor(state, levels = c("raw", "cleaned")))
ggplot(combined_data, aes(x=reorder(sample, sort(as.numeric(sample))), y = sum_len, fill = state)) +
geom_bar(stat = "identity", position = position_dodge(width = 0.8)) +
scale_fill_manual(values = c("raw" = "steelblue", "cleaned" = "firebrick")) +
labs(
x = "Sample",
y = "Length",
fill = "State",
title = "Sum length of reads Before and After Cleaning"
) +
theme_minimal() +
theme(
axis.text.x = element_text(angle = 45, hjust = 1)
)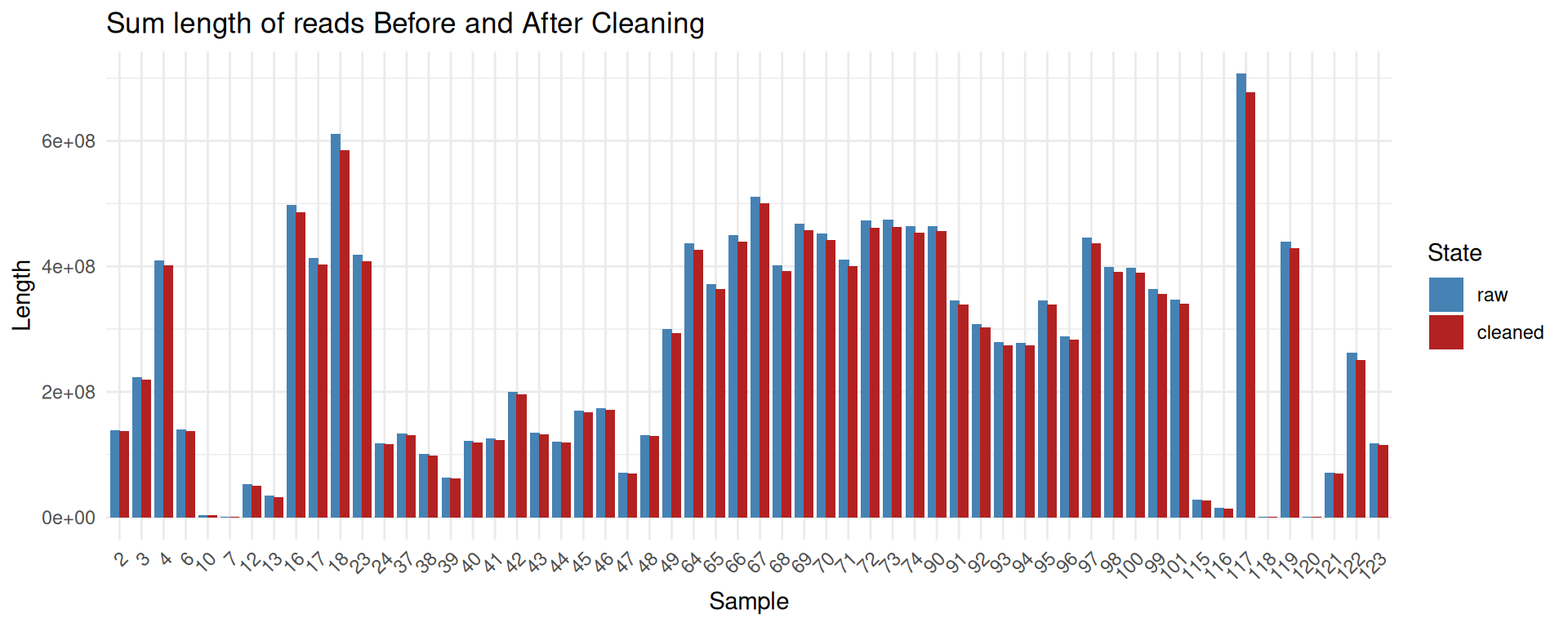
The cleaning process removes only a small fraction of reads and sequenced bases.
cd /home/orue/work/PROJECTS/SCPWI
for s in RAW_DATA/IC_Bre/*.fastq.gz ; do id=$(echo $(basename $s |cut -d '.' -f 1)) ; NanoStat --fastq $s --outdir QC/Breteniere/NANOSTATS/ -n ${id} -t 16 ; donecd /home/orue/work/PROJECTS/SCPWI/REFERENCES
conda activate minimap2-2.28
minimap2 SynCom_Ref16S_Auzeville.fasta -x ava-ont -d SynCom_Ref16S_Auzeville.mmi
minimap2 SynCom_Ref16S_Breteniere.fasta -x ava-ont -d SynCom_Ref16S_Breteniere.mmi
conda activate samtools-1.20 --stack
cd ../MAPPING
mkdir IC_Auz IC_Bre
## IC_Auz
date ; minimap2 -t 96 -ax map-ont -a REFERENCES/GTDB_16S_release220_202411/GTDB_16S_release220_202411.mmi CLEANING/Auzeville/81.fastq.gz |samtools view -bS - |samtools sort -m 16G -@ 96 - > MAPPING/Auzeville/81.bam ; date
for i in ../RAW_DATA/IC_Auz/*.gz ; do id=$(echo $(basename $i) |cut -d '.' -f 1) ; minimap2 -t 24 -ax map-ont -a ../REFERENCES/SynCom_Ref16S_Auzeville.mmi $i |samtools view -bS - |samtools sort - > IC_Auz/${id}.bam ; done
for i in IC_Auz/*.bam ; do samtools flagstat $i > $i.flagstat ; done
for i in IC_Auz/*.bam ; do samtools view $i |cut -f 3 |sort |uniq -c > $i.infos ; done
## IC_Bre
for i in ../RAW_DATA/IC_Bre/*.gz ; do id=$(echo $(basename $i) |cut -d '.' -f 1) ; minimap2 -t 24 -ax map-ont -a ../REFERENCES/SynCom_Ref16S_Breteniere.mmi $i |samtools view -bS - |samtools sort - > IC_Bre/${id}.bam ; done
for i in IC_Bre/*.bam ; do samtools view $i |cut -f 3 |sort |uniq -c > $i.infos ; done
for i in *.bam ; do samtools view -F 2048 $i |cut -f 3 | sort |uniq -c | sort -k1,1nr |perl -lane 'print "$F[0]\t$F[1]"' > ${i}.counts ; done
conda deactivate
for i in CLEANING/Breteniere/*.gz ; do id=$(echo $(basename $i) |cut -d '.' -f 1) ; echo "conda activate minimap2-2.28 && conda activate samtools-1.20 --stack && minimap2 -t 64 -ax map-ont -a REFERENCES/GTDB_16S_release220_202411/GTDB_16S_release220_202411.mmi $i --secondary=no |samtools view -bS - |samtools sort -@ 64 - > MAPPING/Breteniere/${id}.bam" >> mapping_breteniere.sh ; doneAlignment files are filtered on FLAG to keep only the best hit of each read. Then, best hits counts are summed up.
if(!file.exists("html/auzeville.rds")){
file_path <- "data/IC_Auz"
files <- list.files(file_path, pattern = "\\.bam\\.counts$", full.names = TRUE)
data_list <- lapply(files, function(file) {
data <- read.csv2(file, sep="\t", header = FALSE, col.names = c("reads", "GTDB_ID"))
data$Sample <- gsub("\\.bam\\.counts$", "", basename(file))
data <- data %>%
mutate(GTDB_ID = ifelse(GTDB_ID == "*", "unknown", GTDB_ID)) %>%
mutate(Sample = str_trim(Sample)) %>% mutate(Sample = as.character(Sample))
data
})
combined_data <- bind_rows(data_list)
cdata <- combined_data %>% pivot_wider(names_from = Sample, values_from = reads, values_fill = 0)
cdata2 <- left_join(cdata, refs, by="GTDB_ID") %>% separate(Taxonomy, ";(?=[\\S])", into = c("Kingdom","Phylum","Class","Order","Family","Genus","Species"), remove=FALSE) %>% rownames_to_column(var="OTUID") %>% mutate(OTUID = paste0("ASV_",OTUID))
otumat = cdata2 %>% select(-OTUID, -GTDB_ID, -Taxonomy, -Kingdom, -Phylum, -Class, -Order, -Family, -Genus, -Species) %>% as.data.frame()
rownames(otumat) <- cdata2$OTUID
taxmat <- cdata2 %>% select(Kingdom, Phylum, Class, Order, Family, Genus, Species) %>% as.matrix()
rownames(taxmat) <- rownames(otumat)
OTU = otu_table(otumat, taxa_are_rows = TRUE)
TAX = tax_table(taxmat)
physeq = phyloseq(OTU, TAX)
physeq
sample_data(physeq) <- read.table("metadata.tsv", row.names = 1, header = TRUE, sep = "\t", stringsAsFactors = FALSE)
clean_taxon_names <- function(taxon_column) {
gsub("^[a-z]__| \\[id:.*\\]", "", taxon_column)
}
tax_table(physeq) <- apply(tax_table(physeq), 2, clean_taxon_names)
saveRDS(physeq, "html/auzeville.rds")
}else{
physeq <- readRDS("html/auzeville.rds")
}physeqphyloseq-class experiment-level object
otu_table() OTU Table: [ 42016 taxa and 64 samples ]
sample_data() Sample Data: [ 64 samples by 7 sample variables ]
tax_table() Taxonomy Table: [ 42016 taxa by 7 taxonomic ranks ]plot_composition(physeq, "Kingdom", "Bacteria", "Phylum", fill = "Phylum", numberOfTaxa = 20) +
scale_fill_discrete() +
facet_grid(~Compartment, scales = "free_x", space = "free_x")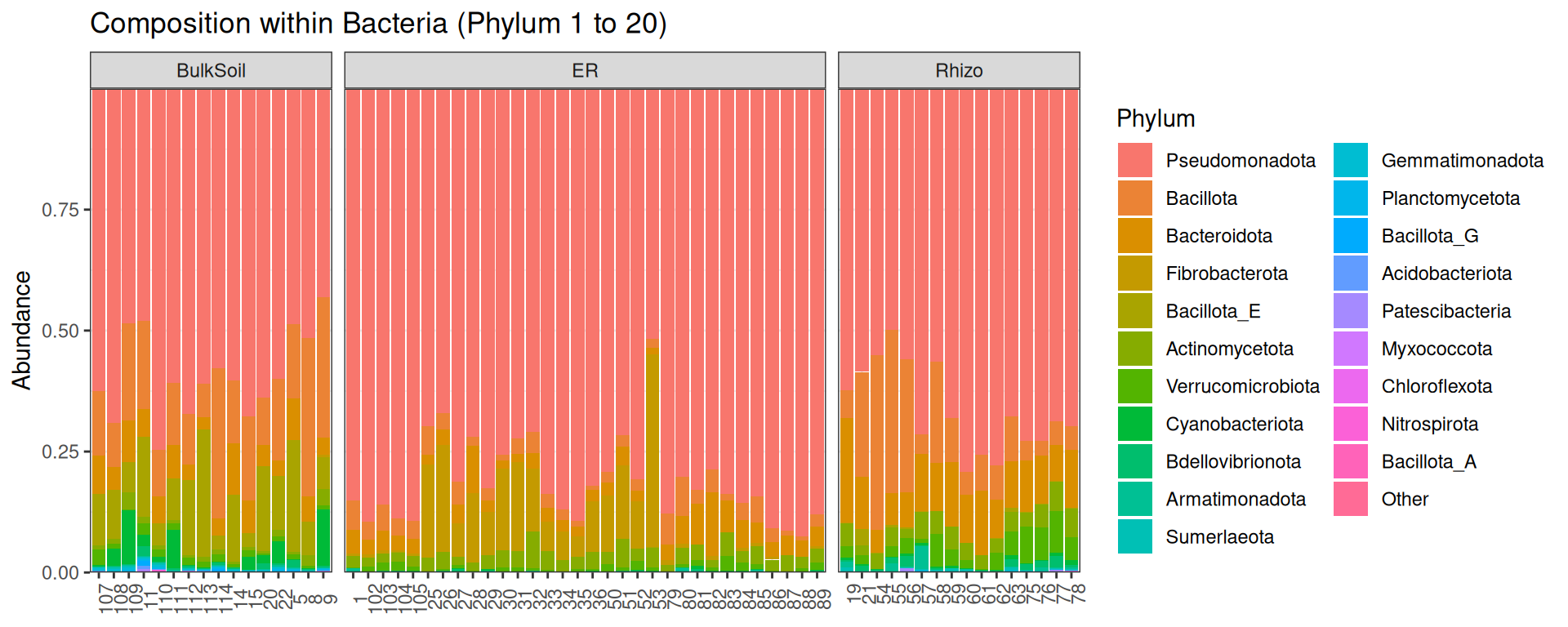
p <- plot_composition(physeq, "Kingdom", "Bacteria", "Species", fill = "Species", numberOfTaxa = 20) +
facet_grid(~Compartment, scales = "free_x", space = "free_x")
p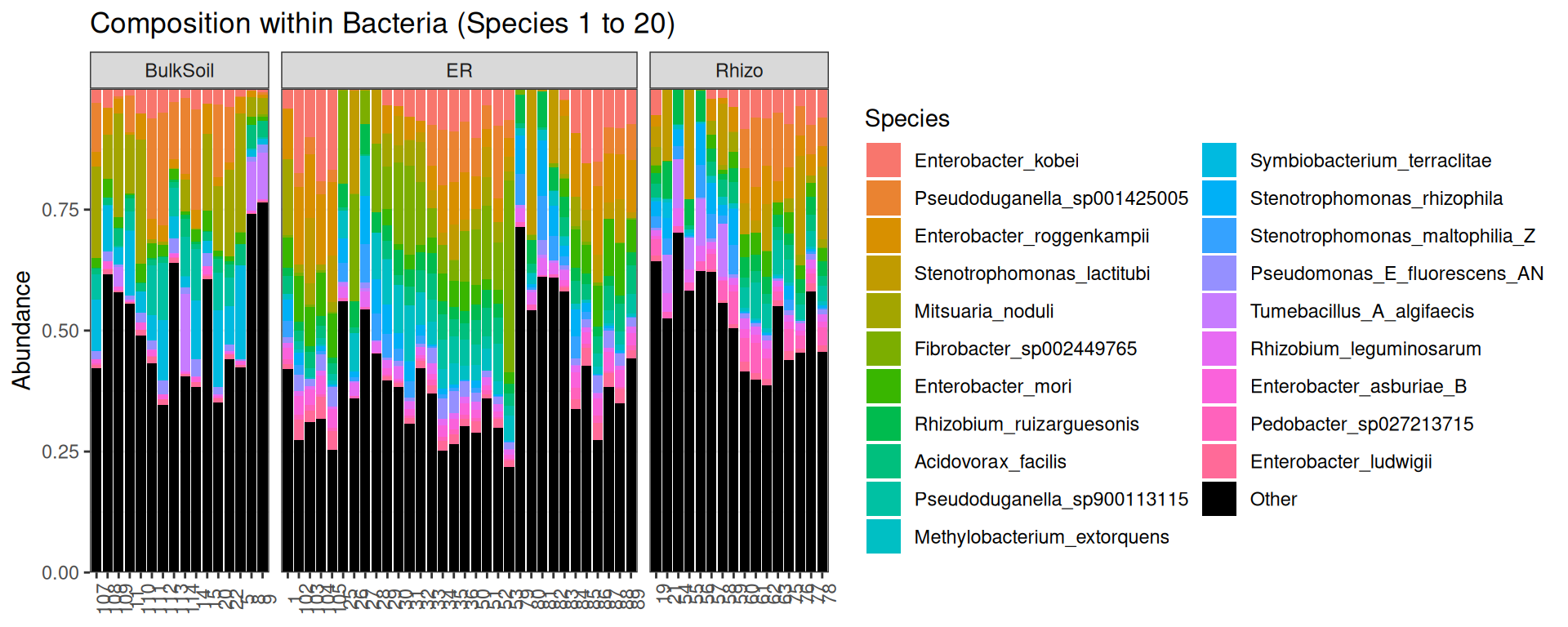
if(!file.exists("html/breteniere.rds")){
file_path <- "data/IC_Bre"
files <- list.files(file_path, pattern = "\\.bam\\.counts$", full.names = TRUE)
data_list <- lapply(files, function(file) {
data <- read.csv2(file, sep="\t", header = FALSE, col.names = c("reads", "GTDB_ID"))
data$Sample <- gsub("\\.bam\\.counts$", "", basename(file))
data <- data %>%
mutate(GTDB_ID = ifelse(GTDB_ID == "*", "unknown", GTDB_ID)) %>%
mutate(Sample = str_trim(Sample)) %>% mutate(Sample = as.character(Sample))
data
})
combined_data <- bind_rows(data_list)
cdata <- combined_data %>% pivot_wider(names_from = Sample, values_from = reads, values_fill = 0)
cdata2 <- left_join(cdata, refs, by="GTDB_ID") %>% separate(Taxonomy, ";(?=[\\S])", into = c("Kingdom","Phylum","Class","Order","Family","Genus","Species"), remove=FALSE) %>% rownames_to_column(var="OTUID") %>% mutate(OTUID = paste0("ASV_",OTUID))
otumat = cdata2 %>% select(-OTUID, -GTDB_ID, -Taxonomy, -Kingdom, -Phylum, -Class, -Order, -Family, -Genus, -Species) %>% as.data.frame()
rownames(otumat) <- cdata2$OTUID
taxmat <- cdata2 %>% select(Kingdom, Phylum, Class, Order, Family, Genus, Species) %>% as.matrix()
rownames(taxmat) <- rownames(otumat)
OTU = otu_table(otumat, taxa_are_rows = TRUE)
TAX = tax_table(taxmat)
physeq = phyloseq(OTU, TAX)
physeq
sample_data(physeq) <- read.table("metadata.tsv", row.names = 1, header = TRUE, sep = "\t", stringsAsFactors = FALSE)
clean_taxon_names <- function(taxon_column) {
gsub("^[a-z]__| \\[id:.*\\]", "", taxon_column)
}
tax_table(physeq) <- apply(tax_table(physeq), 2, clean_taxon_names)
saveRDS(physeq, "html/breteniere.rds")
}else{
physeq <- readRDS("html/breteniere.rds")
}physeqphyloseq-class experiment-level object
otu_table() OTU Table: [ 28026 taxa and 58 samples ]
sample_data() Sample Data: [ 58 samples by 7 sample variables ]
tax_table() Taxonomy Table: [ 28026 taxa by 7 taxonomic ranks ]plot_composition(physeq, "Kingdom", "Bacteria", "Phylum", fill = "Phylum", numberOfTaxa = 20) +
scale_fill_discrete() +
facet_grid(~Compartment, scales = "free_x", space = "free_x")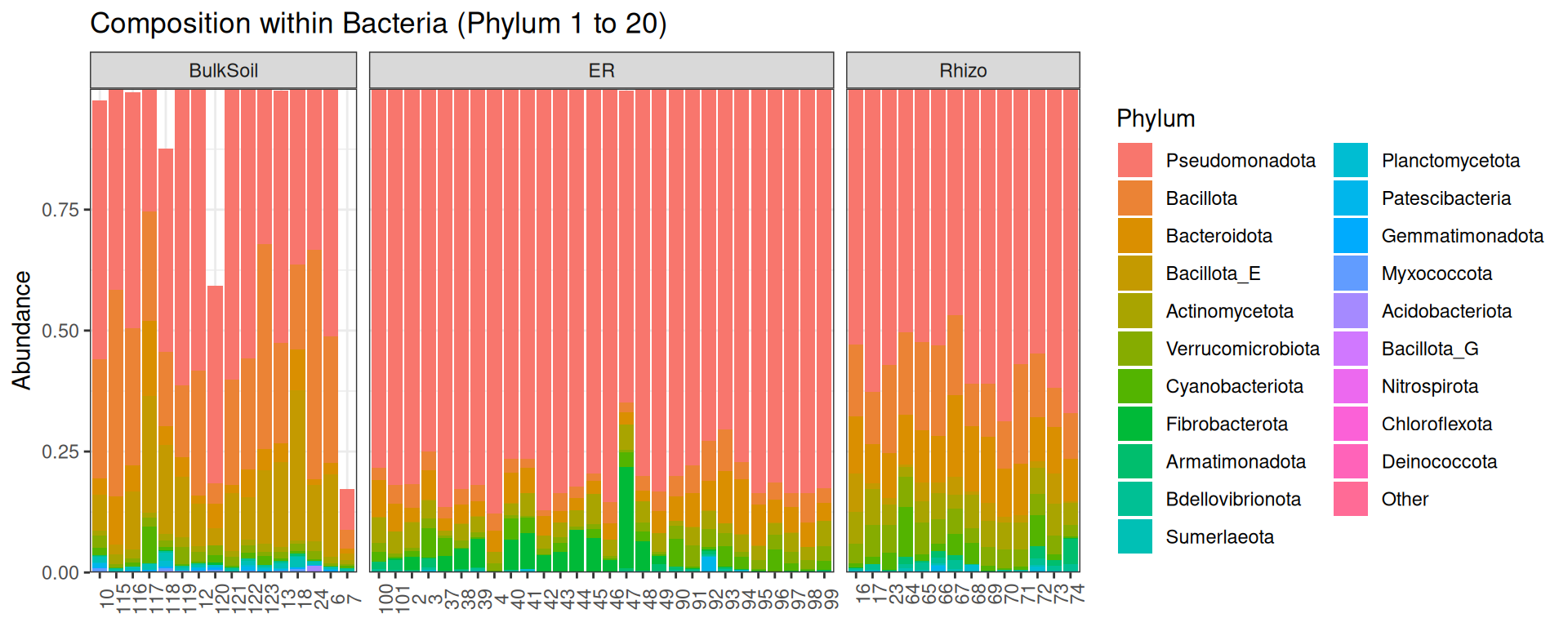
p <- plot_composition(physeq, "Kingdom", "Bacteria", "Species", fill = "Species", numberOfTaxa = 20) +
facet_grid(~Compartment, scales = "free_x", space = "free_x")
p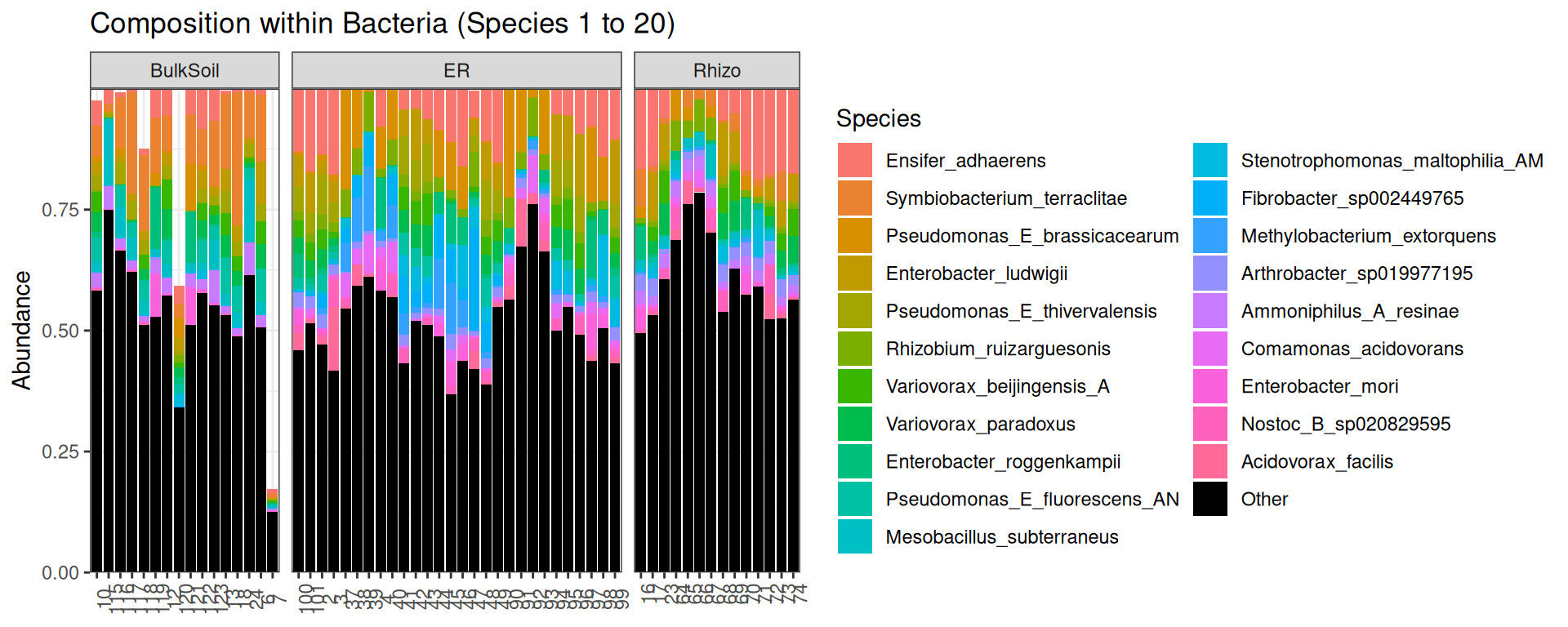
The RDS files can be loaded in the Easy16S application
A work by Migale Bioinformatics Facility
Université Paris-Saclay, INRAE, MaIAGE, 78350, Jouy-en-Josas, France
Université Paris-Saclay, INRAE, BioinfOmics, MIGALE bioinformatics facility, 78350, Jouy-en-Josas, France
