This document is a report of the analyses performed. You will find all the code used to analyze these data. The version of the tools (maybe in code chunks) and their references are indicated, for questions of reproducibility.
Aim of the project
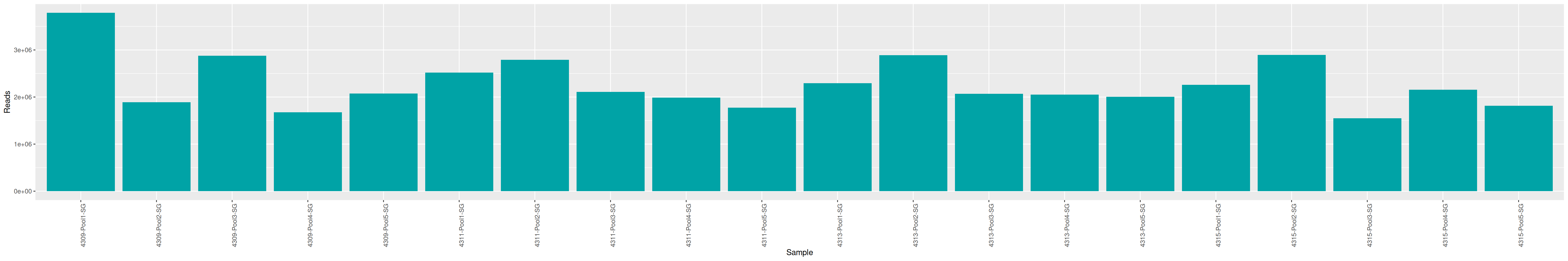
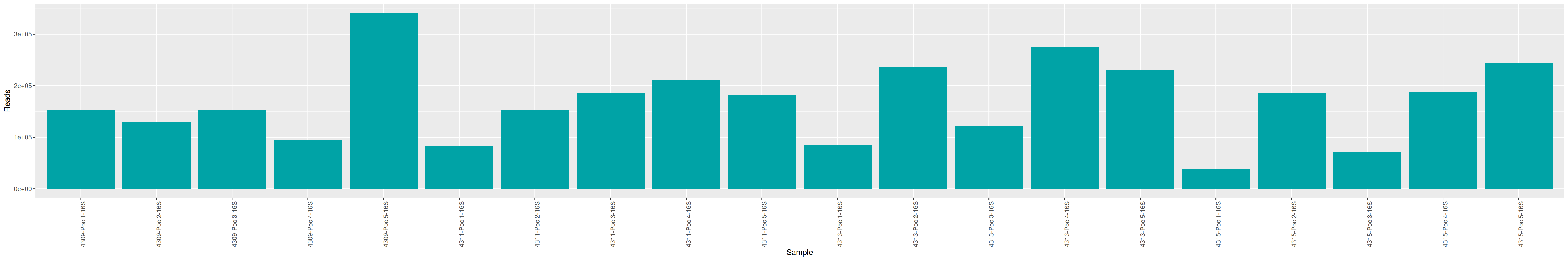
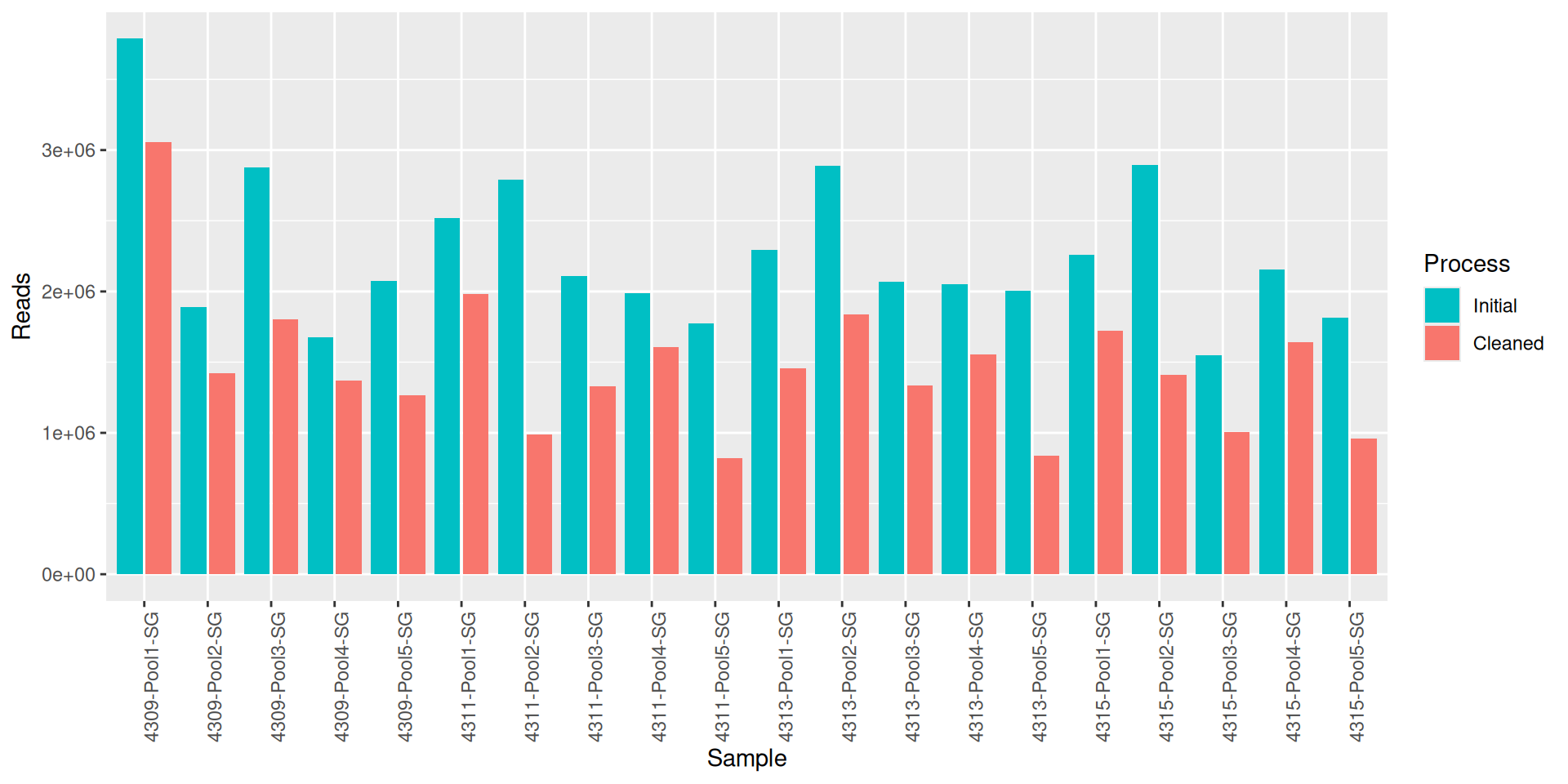
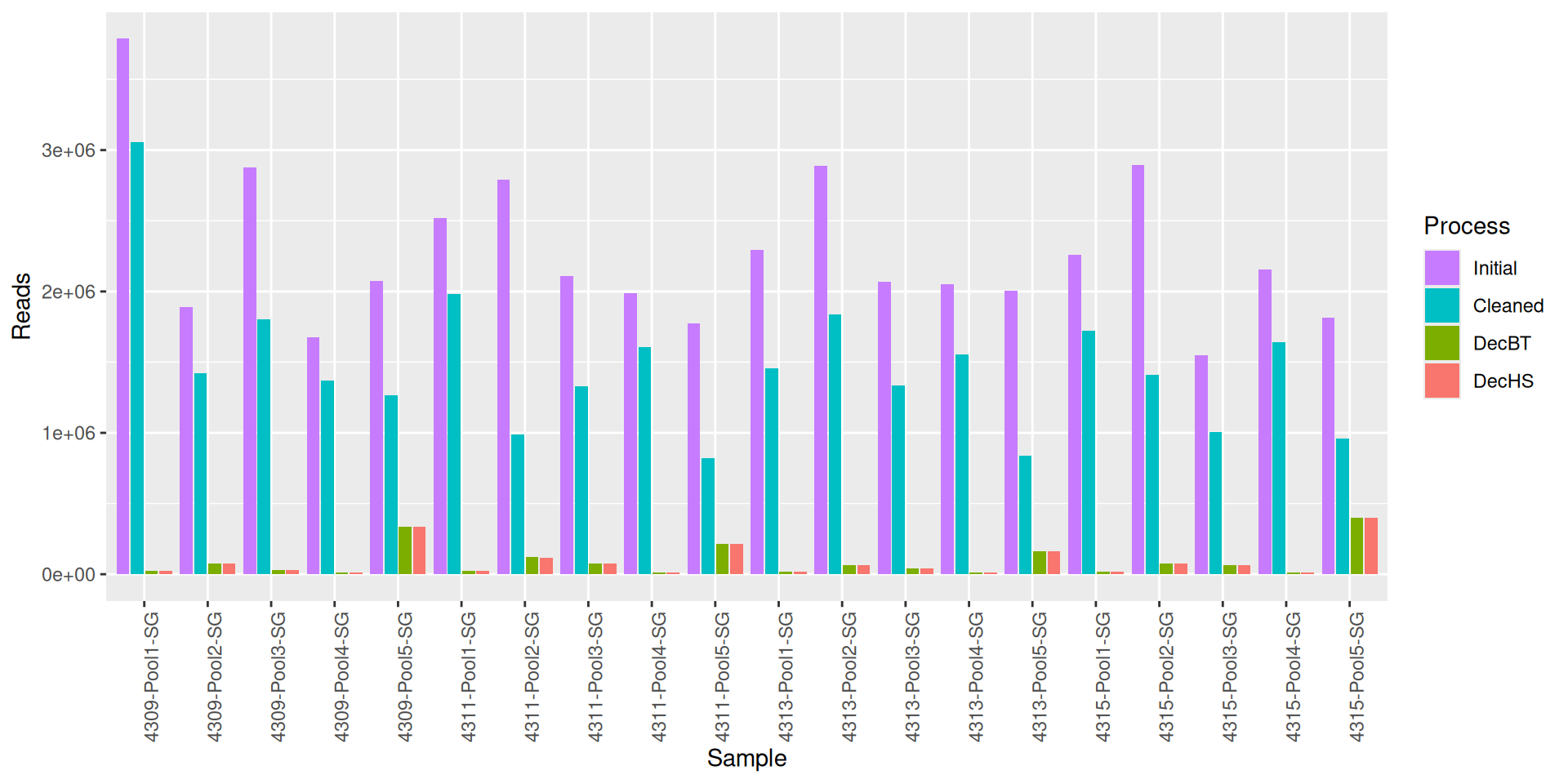
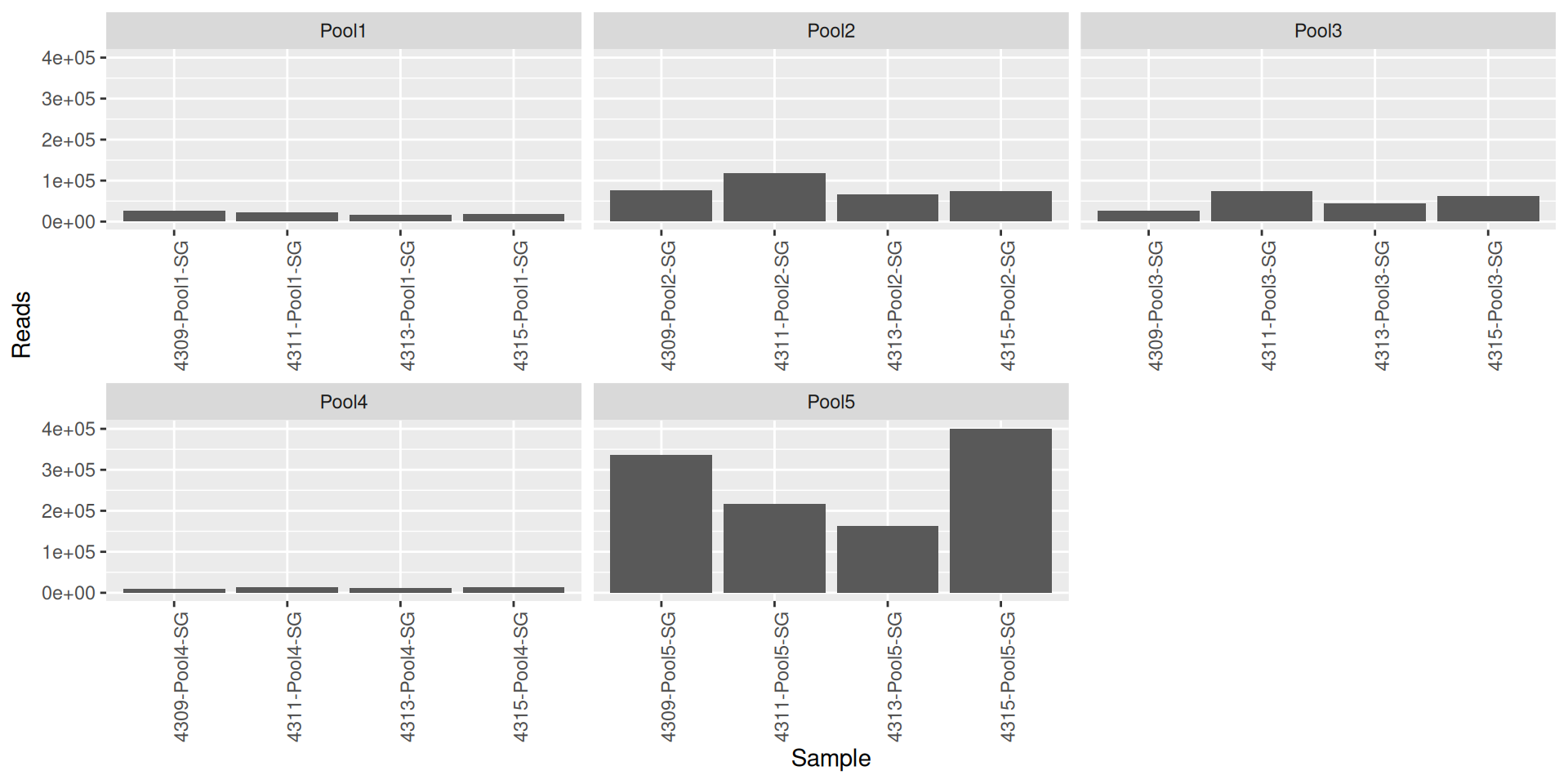
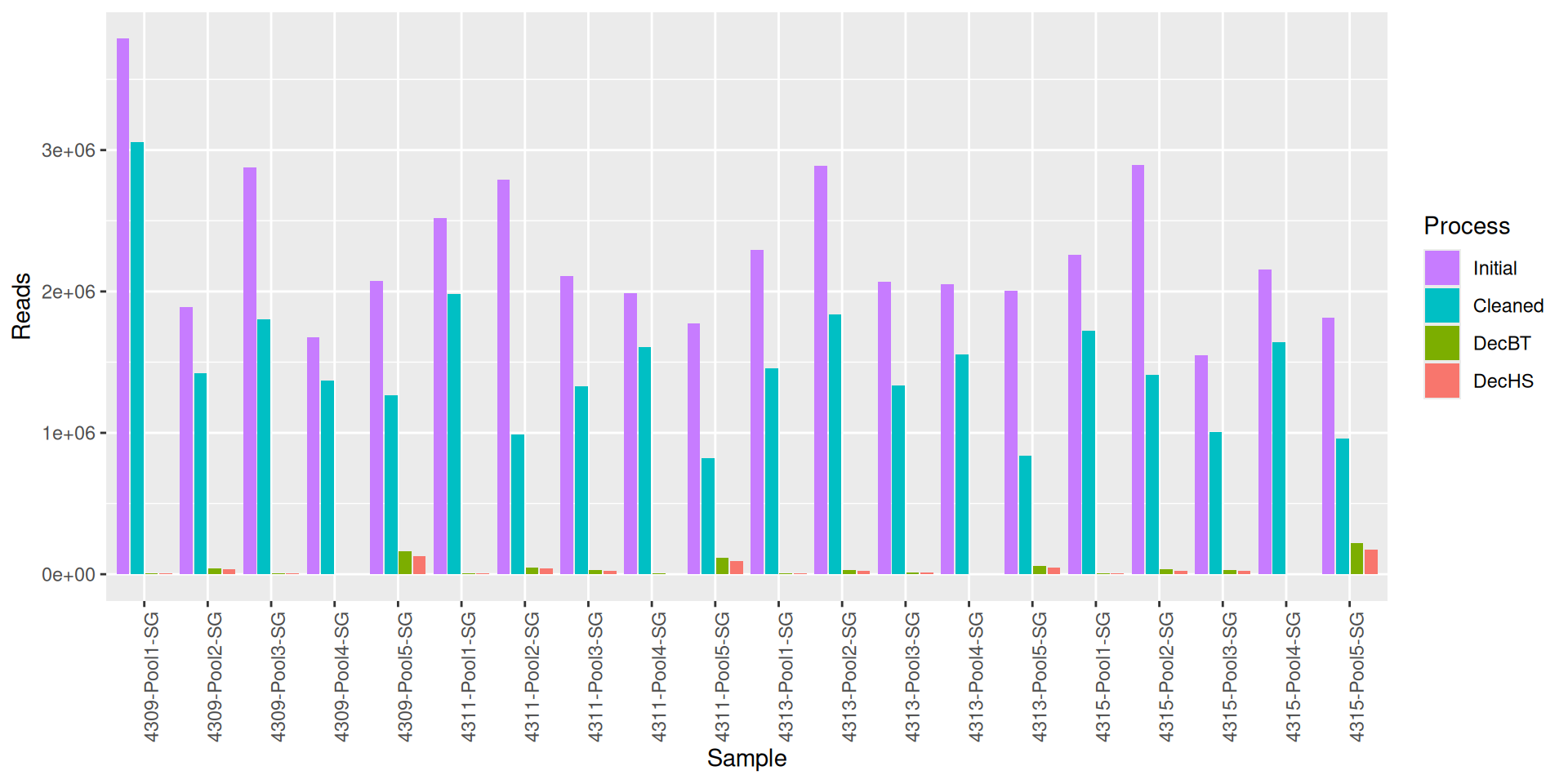
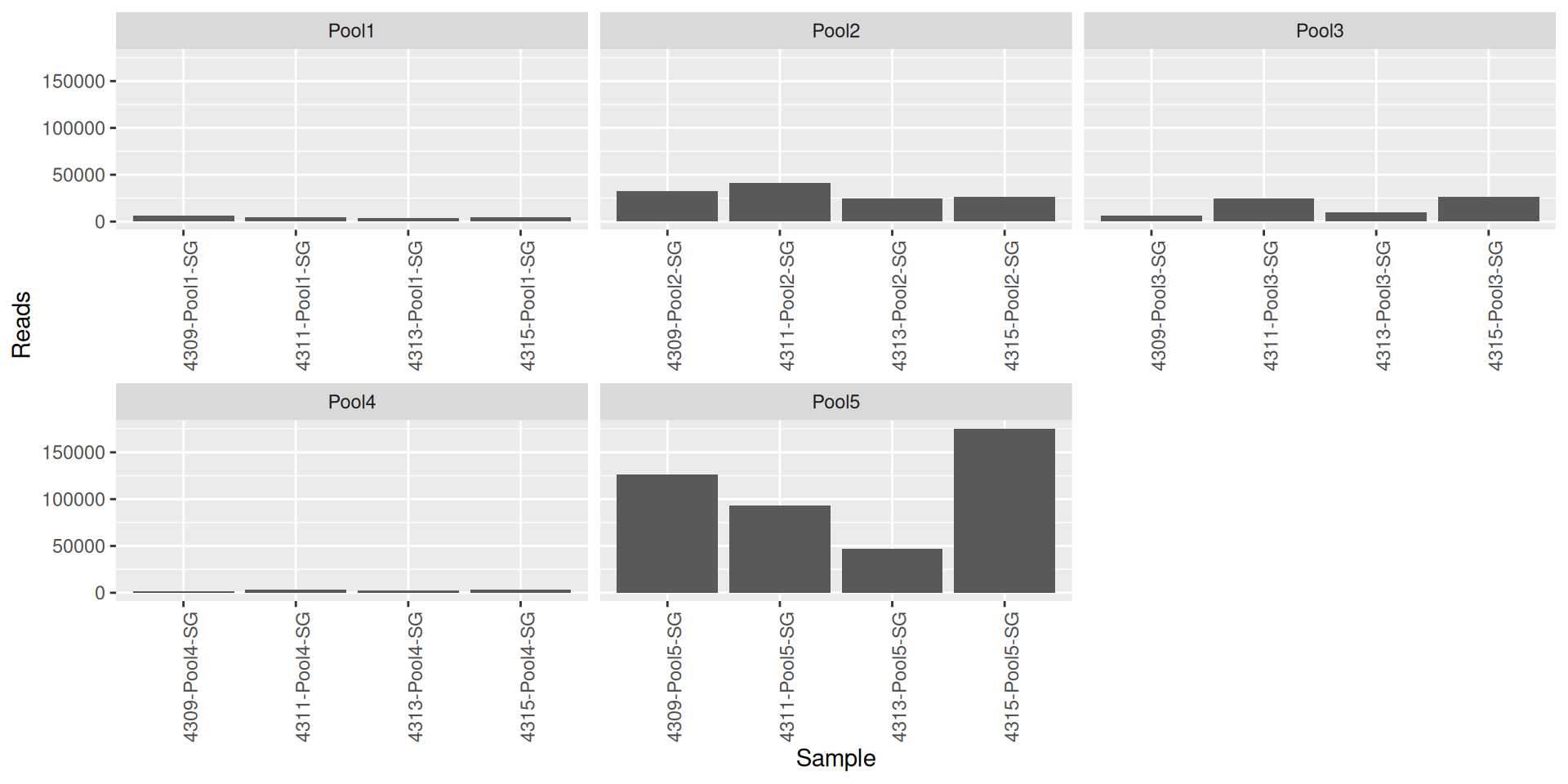
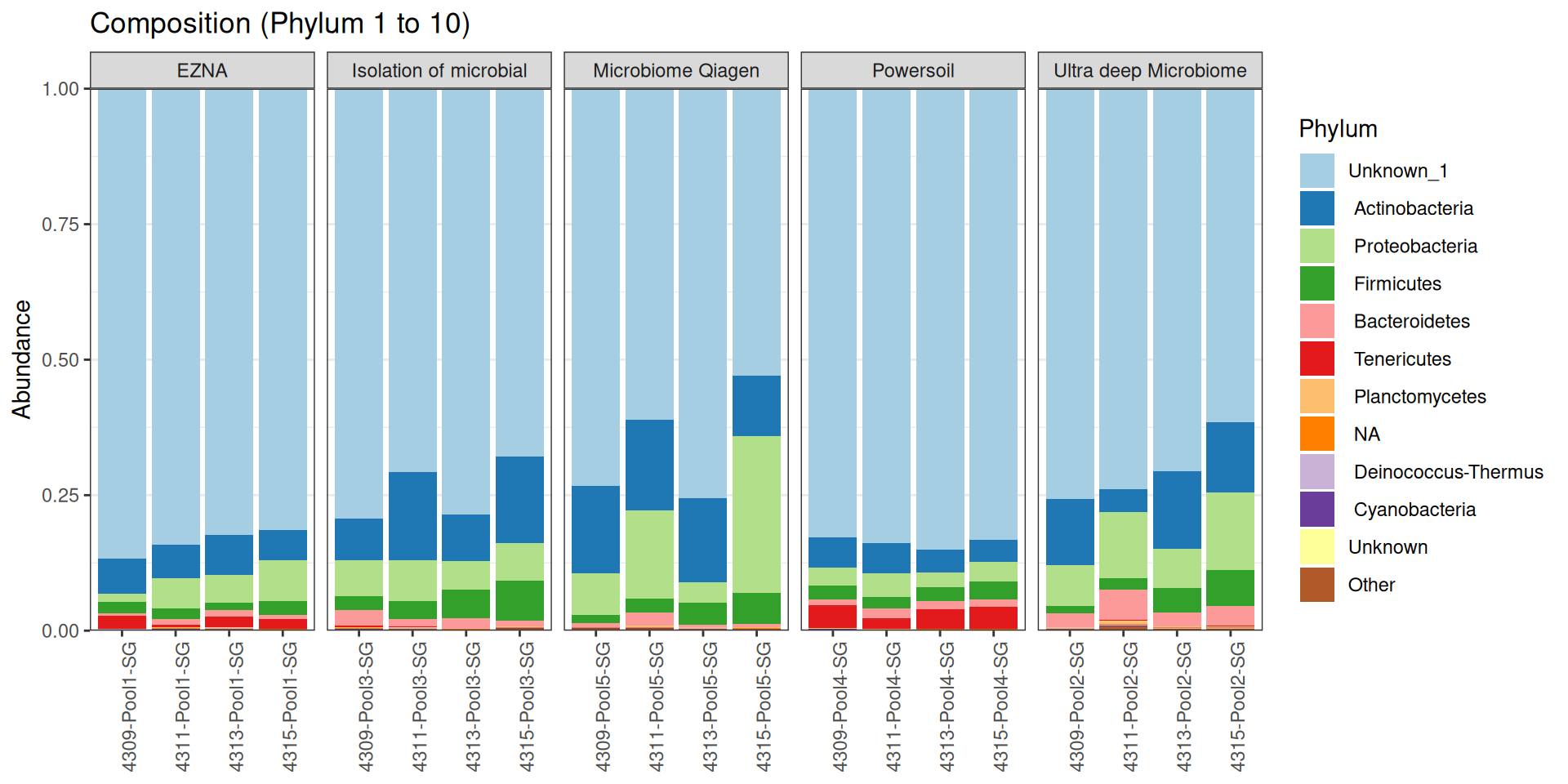
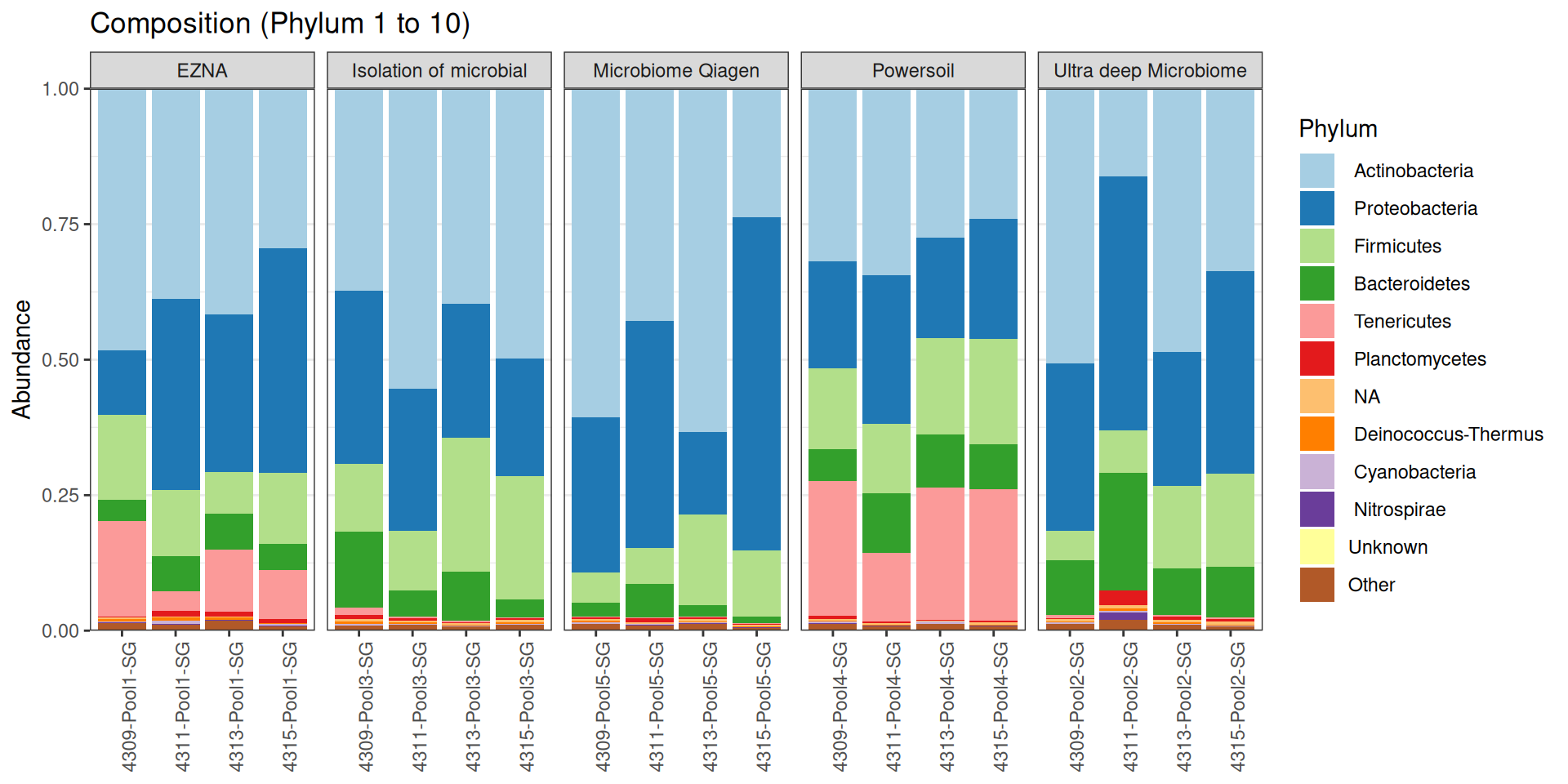
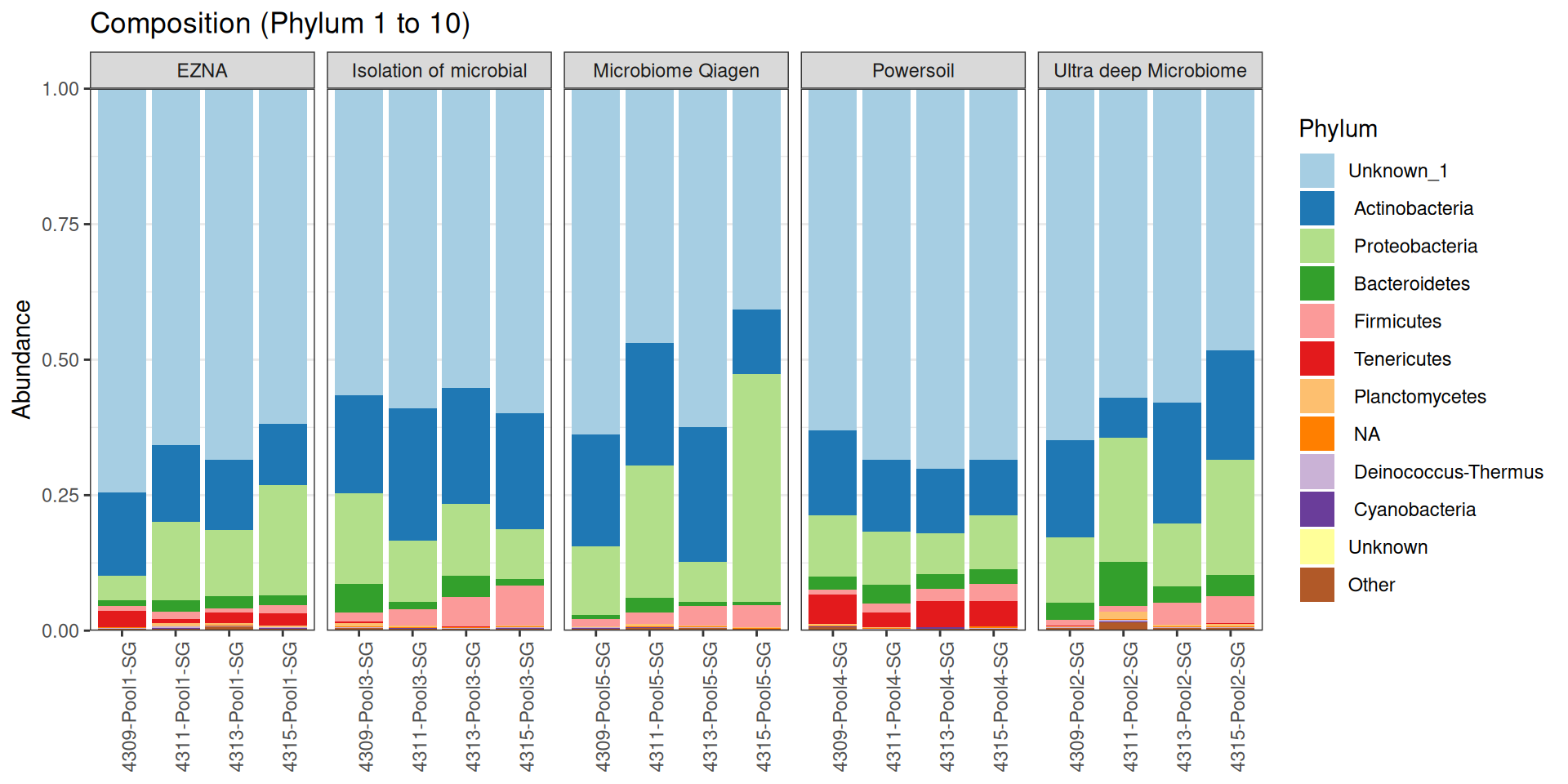
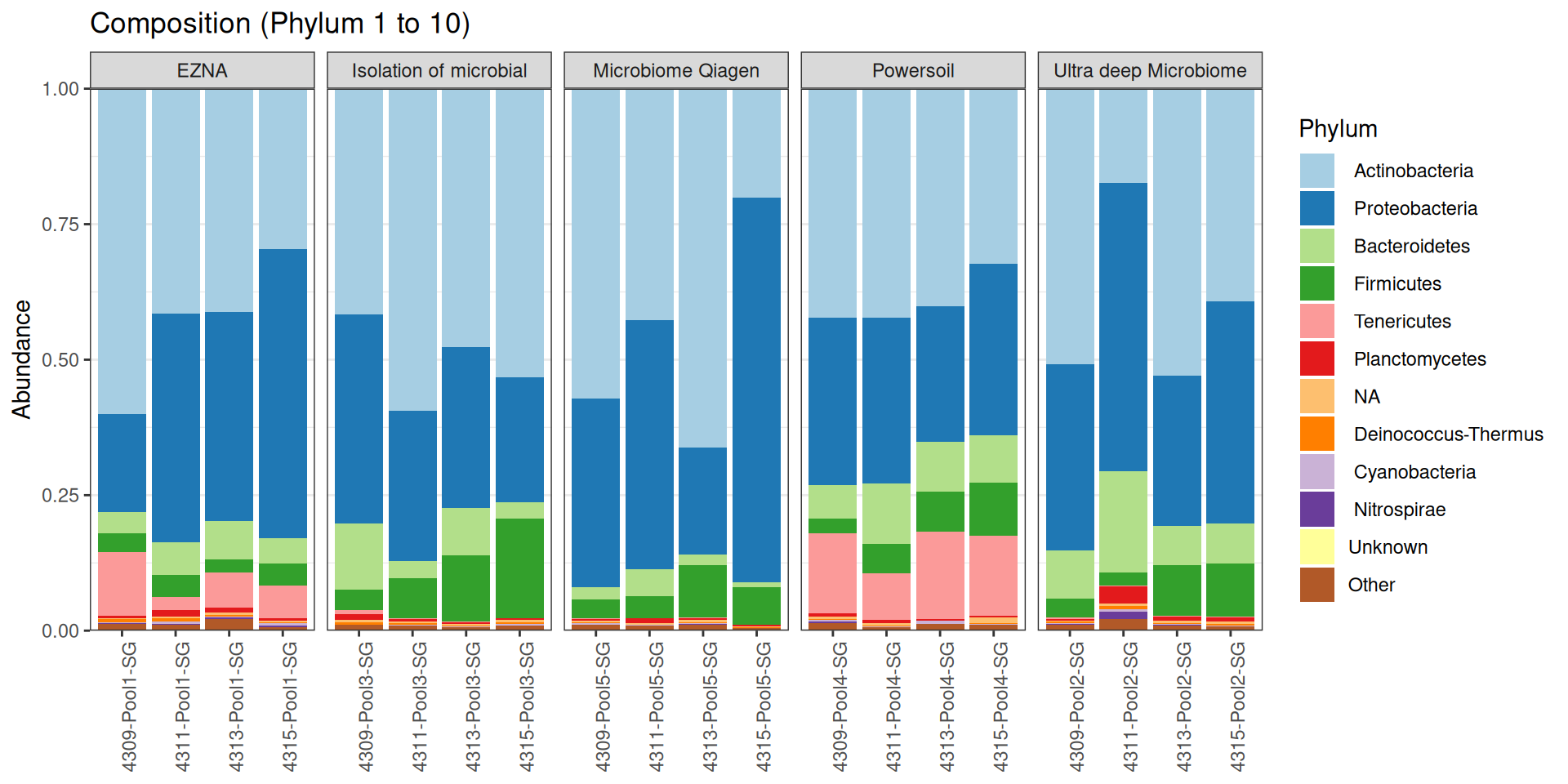
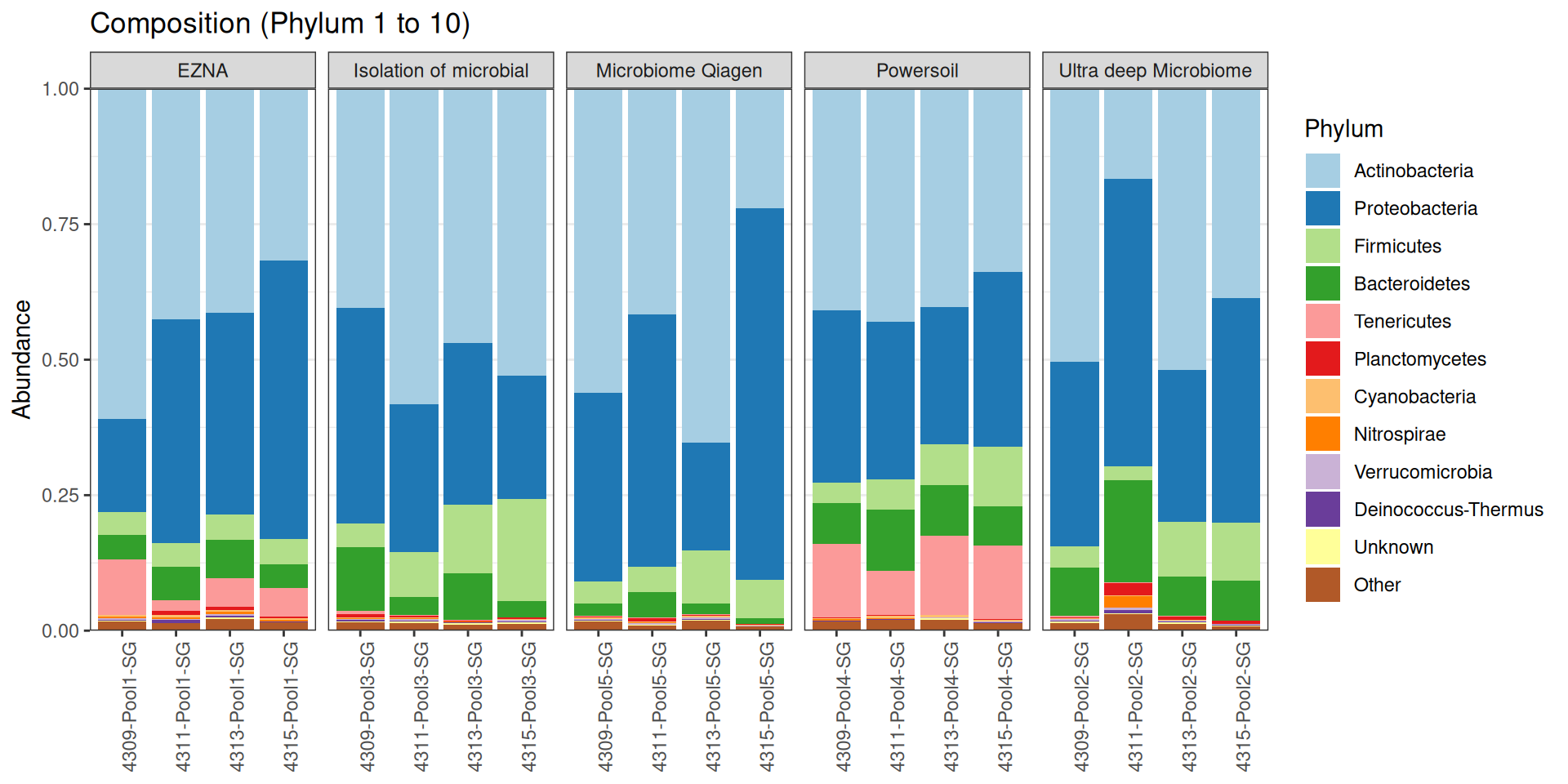
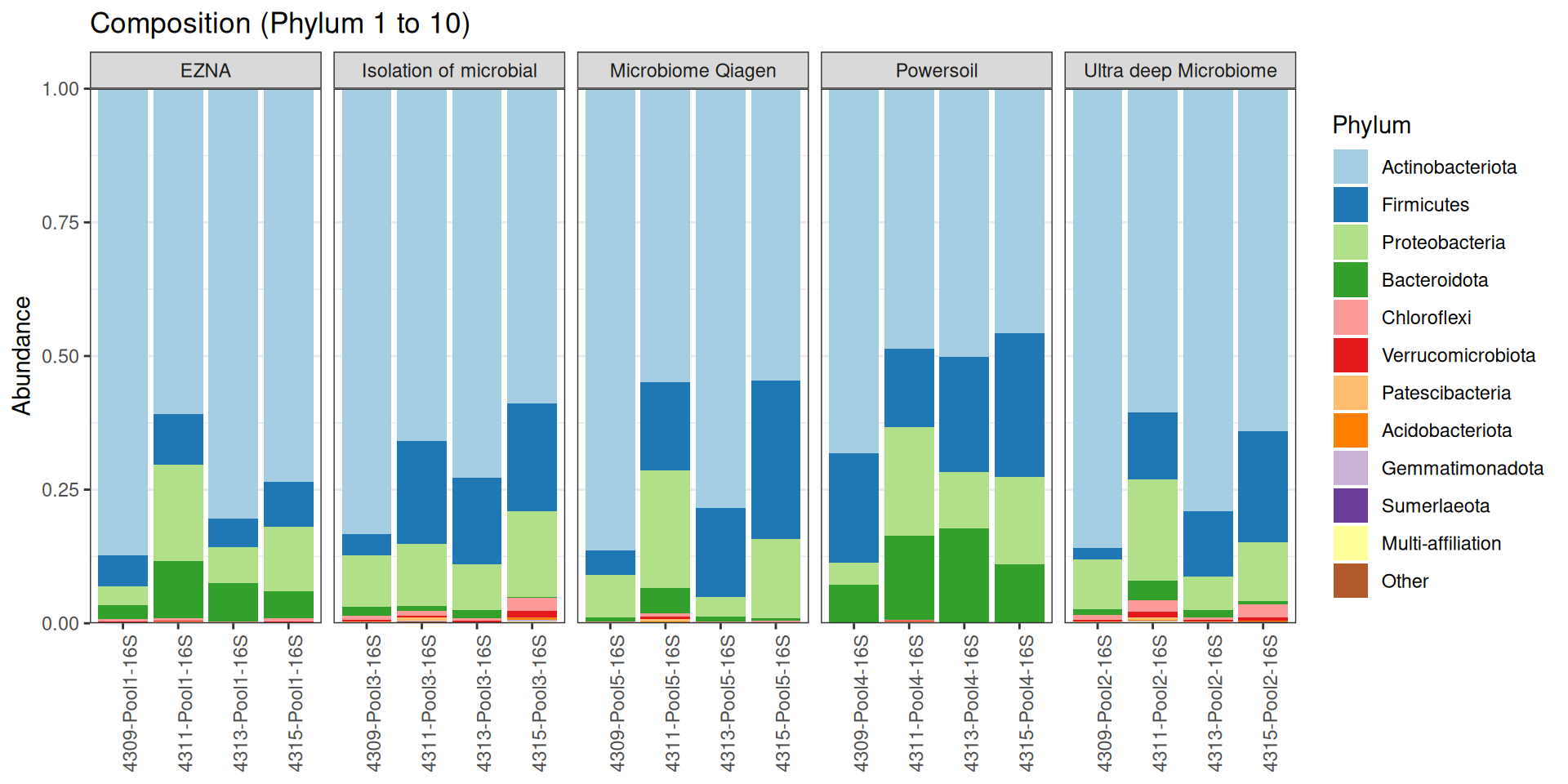
As part of the RESILIENS project, I sent 20 samples for shotgun sequencing to evaluate the efficiency of 5 different DNA extraction kits on 4 respiratory tract samples (4 different animals). The aim of this sequencing was to determine whether the different kits are able to deplete host DNA and thus increase the quantity of microbial reads. To begin with, 2 M reads PE 150bp x sample were requested. The same samples will also be sequenced by 16S sequencing in parallel to get an overview of the composition.
Partners
- Olivier Rué - Migale bioinformatics facility - BioInfomics - INRAE
- Valentin Loux - Migale bioinformatics facility - BioInfomics - INRAE
- Cédric Midoux - Migale bioinformatics facility - BioInfomics - INRAE
- Nuria Mach - UMR 1225 IHAP - INRAE
Deliverables
Deliverables agreed at the preliminary meeting (Table 1).
Data management
All data is managed by the migale facility for the duration of the project. Once the project is over, the Migale facility does not keep your data. We will provide you with the raw data and associated metadata that will be deposited on public repositories before the results are used. We can guide you in the submission process. We will then decide which files to keep, knowing that this report will also be provided to you and that the analyses can be replayed if needed.
Raw data
The raw data were stored in the abaca server and on a dedicated space on the front server.
References
1. Shen W, Le S, Li Y, Hu F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/q file manipulation. PloS one. 2016;11:e0163962.
2. Andrews S. FastQC a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. 2010.
http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
3. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–8.
4. Zhou Y, Chen Y, Chen S, Gu J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–90. doi:
10.1093/bioinformatics/bty560.
5. Rumbavicius I, Rounge TB, Rognes T. HoCoRT: Host contamination removal tool. BMC bioinformatics. 2023;24:371.
6. Menzel P, Ng KL, Krogh A. Fast and sensitive taxonomic classification for metagenomics with kaiju. Nature communications. 2016;7:11257.
7. Escudié F, Auer L, Bernard M, Mariadassou M, Cauquil L, Vidal K, et al.
FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics. 2018;34:1287–94. doi:
10.1093/bioinformatics/btx791.
8. Bernard M, Rué O, Mariadassou M, Pascal G.
FROGS: a powerful tool to analyse the diversity of fungi with special management of internal transcribed spacers. Briefings in Bioinformatics. 2021;22. doi:
10.1093/bib/bbab318.
Reuse
This document will not be accessible without prior agreement of the partners
A work by Migale Bioinformatics Facility
Université Paris-Saclay, INRAE, MaIAGE, 78350, Jouy-en-Josas, France
Université Paris-Saclay, INRAE, BioinfOmics, MIGALE bioinformatics facility, 78350, Jouy-en-Josas, France