
cd /home/orue/work/PROJECTS/OENOVARDOCC2/
mkdir RAW_DATA
find 240426_M03493_0576_000000000-LGBFF/ -name \*.fastq.gz -exec cp {} RAW_DATA/ \;
cd RAW_DATA/
rename 's/_L001//; s/_001//' *.fastq.gz
rename 's/_.*_/_/' *.fastq.gz
rm -f Undete*
scp *.fastq.gz orue@abaca.maiage.inrae.fr:/backup/partage/migale/OENOVARDOCC2/RAW_DATA/OENOVARDOCC2
The aim of this project is to characterize the microbiota of grapes and grape musts from different French vineyards with various grape varieties, 2nd run.
closed
collaboration
Note
This document is a report of the analyses performed. You will find all the code used to analyze these data. The version of the tools (maybe in code chunks) and their references are indicated, for questions of reproducibility.
Aim of the project
The aim of this project is to characterize the microbiota of grapes and grape musts from different French vineyards with various grape varieties. The collect organized in 2023 used 25 different varieties from 4 vineyards. Fermentations were conducted for a limited number of varieties. Samples were collected during the fermentations to investigate the diversity of the microbiota through a metabarcoding approach.
Partners
- Olivier Rué - Migale bioinformatics facility - BioInfomics - INRAE
- Cécile Neuvéglise - SPO - INRAE
Deliverables
Deliverables agreed at the preliminary meeting (Table 1).
| Definition | |
|---|---|
| 1 | HTML report |
Data management
Important
All data is managed by the migale facility for the duration of the project. Once the project is over, the Migale facility does not keep your data. We will provide you with the raw data and associated metadata that will be deposited on public repositories before the results are used. We can guide you in the submission process. We will then decide which files to keep, knowing that this report will also be provided to you and that the analyses can be replayed if needed.
Raw data
Raw data were sent on May 03 and deposited on the front server and a copy was sent to the abaca server.
Quality control
# seqkit
cd /home/orue/work/PROJECTS/OEANOVARDOCC2/
qsub -cwd -V -N seqkit -pe thread 4 -R y -b y "conda activate seqkit-2.0.0 && seqkit stats /home/orue/work/PROJECTS/OENOVARDOCC2/RAW_DATA/*.fastq.gz -j 4 > raw_data.infos && conda deactivate"We can plot and display the number of reads to see if enough reads are present and if samples are homogeneous.
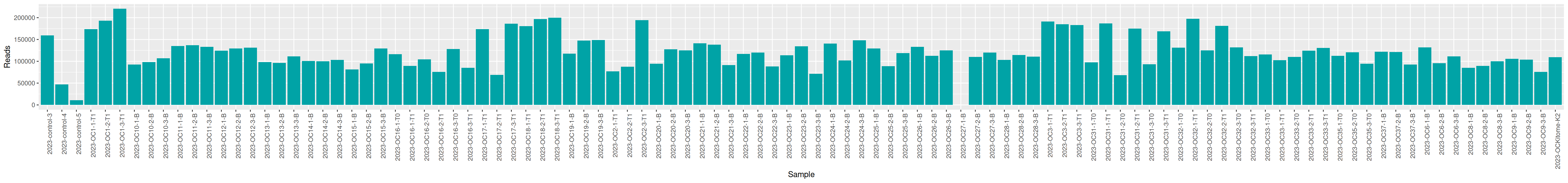
raw_data %>% select(sample, num_seqs, sum_len, min_len, avg_len, max_len) %>% datatable()cd /home/orue/work/PROJECTS/OENOVARDOCC2/
mkdir FASTQC LOGS
for i in /home/orue/work/PROJECTS/OENOVARDOCC2/RAW_DATA/*.fastq.gz ; do echo "conda activate fastqc-0.11.9 && fastqc $i -o FASTQC && conda deactivate" >> fastqc.sh ; done
qarray -cwd -V -N fastqc -o LOGS -e LOGS fastqc.sh
qsub -cwd -V -N multiqc -o LOGS -e LOGS -b y "conda activate multiqc-1.11 && multiqc FASTQC -o MULTIQC && conda deactivate"The MultiQC report shows expected metrics for Illumina Miseq sequencing data.
Note
The quality control is good enough to go further. 1 sample has too few reads: 2023-OC27-1-B and will be discarded at this step.
mkdir TOO_FEW_READS
mv 2023-OC27-1-B_R* TOO_FEW_READS/
tar zcvf Oenovardocc2.tar.gz *.fastq.gzBioinformatics
We used FROGS v.5.0
The first tool, called denoising allows to clean reads. From FASTQ files, reads with N were first discarded. Then, reads were denoised with dada2
cd RAW_DATA_READY
tar zcvf Oenovardocc.tar.gz *.fastq.gz
cd ../
mkdir FROGS5
cd FROGS5
conda activate frogs-5.0.0
denoising.py illumina --min-amplicon-size 50 --max-amplicon-size 1000 --merge-software pear --five-prim-primer CTTGGTCATTTAGAGGAAGTAA --three-prim-primer GCATCGATGAAGAACGCAGC --R1-size 300 --R2-size 300 --nb-cpus 16 --output-fasta clusters.fasta --output-biom clusters.biom --html denoising.html --log-file denoising.log --process dada2 --input-archive ../RAW_DATA/Oenovardocc2.tar.gz --sample-inference pseudo-pooling --keep-unmergedremove_chimera.py --input-biom clusters.biom --input-fasta clusters.fasta --html remove_chimera.html --log-file remove_chimera.log --nb-cpus 16cluster_filters.py --input-fasta remove_chimera.fasta --input-biom remove_chimera_abundance.biom --nb-cpus 16 --contaminant /db/outils/FROGS/contaminants/phi.fa --min-abundance 0.00005 --output-fasta filters.fasta --log-file filters.log --html cluster_filters.htmlitsx.py --check-its-only --input-fasta filters.fasta --input-biom cluster_filters_abundance.biom --log-file itsx.logtaxonomic_affiliation.py --input-biom itsx_abundance.biom --input-fasta itsx.fasta --nb-cpus 16 --reference ~/work/PROJECTS/LEBANESEWHEATSOURDOUGH/ITS/UNITE_9.0_20221016_plus_METABARFOOD.fasta --log-file taxonomic_affiliation.log
Warning
Compared to the metadata file wich contains 106 samples, only 105 samples were present in the sequencing data. The sample 2023-OC27-1-B was removed before bioinformatics (too few reads) but the sample 2023-OCvide was missing.
Correction of taxonomies already changed in previous projects
Now, we will apply changes in taxonomies by comparing ASVs sequences with ASVs manually curated in previous studies (microvarior1,2,3 and oenovardocc1).
First, we build the phyloseq object.
library(Biostrings)
biomfile <- "html/affiliation_abundance.biom"
physeq <- import_frogs(biomfile, taxMethod = "blast")
metadata <- read.table("data/metadata.tsv", row.names = 1, header = TRUE, sep = "\t", stringsAsFactors = FALSE)
metadata <- metadata %>%
mutate(SampleName = rownames(metadata))
sample_data(physeq) <- metadata
fasta_file <- "html/itsx.fasta"
sequences <- readDNAStringSet(fasta_file)
taxa_names(physeq) <- unlist(as.character(sequences))
physeqphyloseq-class experiment-level object
otu_table() OTU Table: [ 488 taxa and 104 samples ]
sample_data() Sample Data: [ 104 samples by 11 sample variables ]
tax_table() Taxonomy Table: [ 488 taxa by 7 taxonomic ranks ]Données Oenovardocc2
Here are the raw taxonomies without any modification:
tibO2 <- psmelt(physeq) %>% as_tibble() %>% mutate(Taxo = paste(Kingdom,Phylum,Class,Order,Family,Genus,Species,sep=";")) %>% group_by(OTU) %>% ungroup() %>% select(OTU,Taxo, Abundance) %>% unique()
datatable(tibO2)Données Oenovardocc1
Here are the ASVs found in Oenovardocc1 project.
oenovardocc1 <- readRDS("html/physeq_curated_with_microvarior3.rds")
fasta_file_oenovardocc1 <- "../oenovardocc-72102/html/itsx.fasta"
sequences_oenovardocc1 <- readDNAStringSet(fasta_file_oenovardocc1)
taxa_names(oenovardocc1) <- unlist(as.character(sequences_oenovardocc1))
asv_oenovardocc1 <- taxa_names(oenovardocc1)
asv_oenovardocc2 <- taxa_names(physeq)
tibO1 <- psmelt(oenovardocc1) %>% as_tibble() %>% mutate(Taxo = paste(Kingdom,Phylum,Class,Order,Family,Genus,Species,sep=";")) %>% group_by(OTU) %>% ungroup() %>% select(OTU,Taxo) %>% unique()
datatable(tibO1)We will change taxonomies for similar ASVs sequence if a taxonomy is present in previous studies.
library(phyloseq)
library(dplyr)
# Fonction pour mettre à jour la taxonomie d'un objet phyloseq avec celle d'un autre
update_taxonomy <- function(physeq_target, physeq_source) {
# Extraire les tables de taxonomie
tax_target <- as.data.frame(tax_table(physeq_target))
tax_source <- as.data.frame(tax_table(physeq_source))
# Ajouter les noms de taxa (séquences ASV) comme colonne
tax_target$ASV <- rownames(tax_target)
tax_source$ASV <- rownames(tax_source)
# Effectuer la jointure sur les noms de taxa (séquences ASV) avec des correspondances exactes
updated_tax <- tax_target %>%
left_join(tax_source, by = "ASV", suffix = c(".target", ".source"))
# Garder seulement les colonnes source (les nouvelles taxonomies)
# Utiliser les colonnes target si les colonnes source sont manquantes
updated_tax <- updated_tax %>%
mutate(across(ends_with(".source"), ~ coalesce(., get(sub(".source", ".target", cur_column()))))) %>%
select(ASV, ends_with(".source"))
# Supprimer les suffixes des colonnes
colnames(updated_tax) <- gsub("\\.source", "", colnames(updated_tax))
# Mettre à jour la table de taxonomie du premier objet phyloseq
new_tax_table <- as.matrix(updated_tax[, -1])
rownames(new_tax_table) <- updated_tax$ASV
tax_table(physeq_target) <- tax_table(new_tax_table)
# Vérifier et mettre à jour les noms de taxa dans tous les composants de l'objet phyloseq
taxa_names(physeq_target) <- updated_tax$ASV
if (!is.null(otu_table(physeq_target))) {
taxa_names(otu_table(physeq_target)) <- updated_tax$ASV
}
#if (!is.null(phy_tree(physeq_target))) {
# taxa_names(phy_tree(physeq_target)) <- updated_tax$ASV
#}
if (!is.null(tax_table(physeq_target))) {
taxa_names(tax_table(physeq_target)) <- updated_tax$ASV
}
return(physeq_target)
}
# Exemple d'utilisation
# Supposons que physeq1 est le phyloseq cible et physeq2 est le phyloseq source
physeq_updated <- update_taxonomy(physeq, oenovardocc1)p <- plot_composition(physeq = physeq_updated, taxaRank1 = "Kingdom", taxaSet1 = "Fungi", taxaRank2 = "Family", numberOfTaxa = 20L, spread = TRUE) Problematic taxa
taxa
AAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAGTAAGGGTGCTCAGCGCCCGACCTCCAACCCTTTGTTGTTAAAACTACCTTGTTGCTTTGGCGGGACCGCTCGGTCTCGAGCCGCTGGGGATTCGTCCCAGGCGAGCGCCCGCCAGAGTTAAACCAAACTCTTGTTATTTAACCGGTCGTCTGAGTTAAAATTTTGAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTC AAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAGTAAGGGTGCTCAGCGCCCGACCTCCAACCCTTTGTTGTTAAAACTACCTTGTTGCTTTGGCGGGACCGCTCGGTCTCGAGCCGCTGGGGATTCGTCCCAGGCGAGCGCCCGCCAGAGTTAAACCAAACTCTTGTTATTTAACCGGTCGTCTGAGTTAAAATTTTGAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTC
AAGTCGTAACAAGGTCTCCGTAGGTGAACCTGCGGAGGGATCATTACACAATATGAAAGCGGGTTGGGACCTCACCTCGGTGAGGGCTCCAGCTTGTCTGAATTATTCACCCATGTCTTTTGCGCACTTCTTGTTTCCTGGGCGGGTTCGCCCGCCACCAGGACCAAACCATAAACCTTTTTGTAATTGCAATCAGCGTCAGTAAACAATGTAATTATTACAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCANNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNTTGGTCATTTAGAGGAAGTAAAAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAGTAAGGGTGCTCAGCGCCCGACCTCCAACCCTTTGTTGTTAAAACTACCTTGTTGCTTTGGCGGGACCGCTCGGTCTCGAGCCGCTGGGGATTCGTCCCAGGCGAGCGCCCGCCAGAGTTAAACCAAACTCTTGTTATTTAACCGGTCGTCTGAGTTAAAATTTTGAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTC AAGTCGTAACAAGGTCTCCGTAGGTGAACCTGCGGAGGGATCATTACACAATATGAAAGCGGGTTGGGACCTCACCTCGGTGAGGGCTCCAGCTTGTCTGAATTATTCACCCATGTCTTTTGCGCACTTCTTGTTTCCTGGGCGGGTTCGCCCGCCACCAGGACCAAACCATAAACCTTTTTGTAATTGCAATCAGCGTCAGTAAACAATGTAATTATTACAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCANNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNTTGGTCATTTAGAGGAAGTAAAAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAGTAAGGGTGCTCAGCGCCCGACCTCCAACCCTTTGTTGTTAAAACTACCTTGTTGCTTTGGCGGGACCGCTCGGTCTCGAGCCGCTGGGGATTCGTCCCAGGCGAGCGCCCGCCAGAGTTAAACCAAACTCTTGTTATTTAACCGGTCGTCTGAGTTAAAATTTTGAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTC
Kingdom
AAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAGTAAGGGTGCTCAGCGCCCGACCTCCAACCCTTTGTTGTTAAAACTACCTTGTTGCTTTGGCGGGACCGCTCGGTCTCGAGCCGCTGGGGATTCGTCCCAGGCGAGCGCCCGCCAGAGTTAAACCAAACTCTTGTTATTTAACCGGTCGTCTGAGTTAAAATTTTGAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTC Fungi
AAGTCGTAACAAGGTCTCCGTAGGTGAACCTGCGGAGGGATCATTACACAATATGAAAGCGGGTTGGGACCTCACCTCGGTGAGGGCTCCAGCTTGTCTGAATTATTCACCCATGTCTTTTGCGCACTTCTTGTTTCCTGGGCGGGTTCGCCCGCCACCAGGACCAAACCATAAACCTTTTTGTAATTGCAATCAGCGTCAGTAAACAATGTAATTATTACAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCANNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNTTGGTCATTTAGAGGAAGTAAAAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAGTAAGGGTGCTCAGCGCCCGACCTCCAACCCTTTGTTGTTAAAACTACCTTGTTGCTTTGGCGGGACCGCTCGGTCTCGAGCCGCTGGGGATTCGTCCCAGGCGAGCGCCCGCCAGAGTTAAACCAAACTCTTGTTATTTAACCGGTCGTCTGAGTTAAAATTTTGAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTC Fungi
Phylum
AAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAGTAAGGGTGCTCAGCGCCCGACCTCCAACCCTTTGTTGTTAAAACTACCTTGTTGCTTTGGCGGGACCGCTCGGTCTCGAGCCGCTGGGGATTCGTCCCAGGCGAGCGCCCGCCAGAGTTAAACCAAACTCTTGTTATTTAACCGGTCGTCTGAGTTAAAATTTTGAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTC Ascomycota
AAGTCGTAACAAGGTCTCCGTAGGTGAACCTGCGGAGGGATCATTACACAATATGAAAGCGGGTTGGGACCTCACCTCGGTGAGGGCTCCAGCTTGTCTGAATTATTCACCCATGTCTTTTGCGCACTTCTTGTTTCCTGGGCGGGTTCGCCCGCCACCAGGACCAAACCATAAACCTTTTTGTAATTGCAATCAGCGTCAGTAAACAATGTAATTATTACAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCANNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNTTGGTCATTTAGAGGAAGTAAAAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAGTAAGGGTGCTCAGCGCCCGACCTCCAACCCTTTGTTGTTAAAACTACCTTGTTGCTTTGGCGGGACCGCTCGGTCTCGAGCCGCTGGGGATTCGTCCCAGGCGAGCGCCCGCCAGAGTTAAACCAAACTCTTGTTATTTAACCGGTCGTCTGAGTTAAAATTTTGAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTC Ascomycota
Class
AAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAGTAAGGGTGCTCAGCGCCCGACCTCCAACCCTTTGTTGTTAAAACTACCTTGTTGCTTTGGCGGGACCGCTCGGTCTCGAGCCGCTGGGGATTCGTCCCAGGCGAGCGCCCGCCAGAGTTAAACCAAACTCTTGTTATTTAACCGGTCGTCTGAGTTAAAATTTTGAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTC Dothideomycetes
AAGTCGTAACAAGGTCTCCGTAGGTGAACCTGCGGAGGGATCATTACACAATATGAAAGCGGGTTGGGACCTCACCTCGGTGAGGGCTCCAGCTTGTCTGAATTATTCACCCATGTCTTTTGCGCACTTCTTGTTTCCTGGGCGGGTTCGCCCGCCACCAGGACCAAACCATAAACCTTTTTGTAATTGCAATCAGCGTCAGTAAACAATGTAATTATTACAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCANNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNTTGGTCATTTAGAGGAAGTAAAAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAGTAAGGGTGCTCAGCGCCCGACCTCCAACCCTTTGTTGTTAAAACTACCTTGTTGCTTTGGCGGGACCGCTCGGTCTCGAGCCGCTGGGGATTCGTCCCAGGCGAGCGCCCGCCAGAGTTAAACCAAACTCTTGTTATTTAACCGGTCGTCTGAGTTAAAATTTTGAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTC Saccharomycetes
Order
AAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAGTAAGGGTGCTCAGCGCCCGACCTCCAACCCTTTGTTGTTAAAACTACCTTGTTGCTTTGGCGGGACCGCTCGGTCTCGAGCCGCTGGGGATTCGTCCCAGGCGAGCGCCCGCCAGAGTTAAACCAAACTCTTGTTATTTAACCGGTCGTCTGAGTTAAAATTTTGAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTC Dothideales
AAGTCGTAACAAGGTCTCCGTAGGTGAACCTGCGGAGGGATCATTACACAATATGAAAGCGGGTTGGGACCTCACCTCGGTGAGGGCTCCAGCTTGTCTGAATTATTCACCCATGTCTTTTGCGCACTTCTTGTTTCCTGGGCGGGTTCGCCCGCCACCAGGACCAAACCATAAACCTTTTTGTAATTGCAATCAGCGTCAGTAAACAATGTAATTATTACAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCANNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNTTGGTCATTTAGAGGAAGTAAAAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAGTAAGGGTGCTCAGCGCCCGACCTCCAACCCTTTGTTGTTAAAACTACCTTGTTGCTTTGGCGGGACCGCTCGGTCTCGAGCCGCTGGGGATTCGTCCCAGGCGAGCGCCCGCCAGAGTTAAACCAAACTCTTGTTATTTAACCGGTCGTCTGAGTTAAAATTTTGAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTC Dothideales
Family
AAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAGTAAGGGTGCTCAGCGCCCGACCTCCAACCCTTTGTTGTTAAAACTACCTTGTTGCTTTGGCGGGACCGCTCGGTCTCGAGCCGCTGGGGATTCGTCCCAGGCGAGCGCCCGCCAGAGTTAAACCAAACTCTTGTTATTTAACCGGTCGTCTGAGTTAAAATTTTGAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTC Dothioraceae
AAGTCGTAACAAGGTCTCCGTAGGTGAACCTGCGGAGGGATCATTACACAATATGAAAGCGGGTTGGGACCTCACCTCGGTGAGGGCTCCAGCTTGTCTGAATTATTCACCCATGTCTTTTGCGCACTTCTTGTTTCCTGGGCGGGTTCGCCCGCCACCAGGACCAAACCATAAACCTTTTTGTAATTGCAATCAGCGTCAGTAAACAATGTAATTATTACAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCANNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNTTGGTCATTTAGAGGAAGTAAAAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAGTAAGGGTGCTCAGCGCCCGACCTCCAACCCTTTGTTGTTAAAACTACCTTGTTGCTTTGGCGGGACCGCTCGGTCTCGAGCCGCTGGGGATTCGTCCCAGGCGAGCGCCCGCCAGAGTTAAACCAAACTCTTGTTATTTAACCGGTCGTCTGAGTTAAAATTTTGAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTC Dothioraceae
rank
AAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAGTAAGGGTGCTCAGCGCCCGACCTCCAACCCTTTGTTGTTAAAACTACCTTGTTGCTTTGGCGGGACCGCTCGGTCTCGAGCCGCTGGGGATTCGTCCCAGGCGAGCGCCCGCCAGAGTTAAACCAAACTCTTGTTATTTAACCGGTCGTCTGAGTTAAAATTTTGAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTC 6
AAGTCGTAACAAGGTCTCCGTAGGTGAACCTGCGGAGGGATCATTACACAATATGAAAGCGGGTTGGGACCTCACCTCGGTGAGGGCTCCAGCTTGTCTGAATTATTCACCCATGTCTTTTGCGCACTTCTTGTTTCCTGGGCGGGTTCGCCCGCCACCAGGACCAAACCATAAACCTTTTTGTAATTGCAATCAGCGTCAGTAAACAATGTAATTATTACAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCANNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNTTGGTCATTTAGAGGAAGTAAAAGTCGTAACAAGGTTTCCGTAGGTGAACCTGCGGAAGGATCATTAAAGAGTAAGGGTGCTCAGCGCCCGACCTCCAACCCTTTGTTGTTAAAACTACCTTGTTGCTTTGGCGGGACCGCTCGGTCTCGAGCCGCTGGGGATTCGTCCCAGGCGAGCGCCCGCCAGAGTTAAACCAAACTCTTGTTATTTAACCGGTCGTCTGAGTTAAAATTTTGAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTC 17p+ facet_grid(~Variety_name, scales = "free_x", space = "free")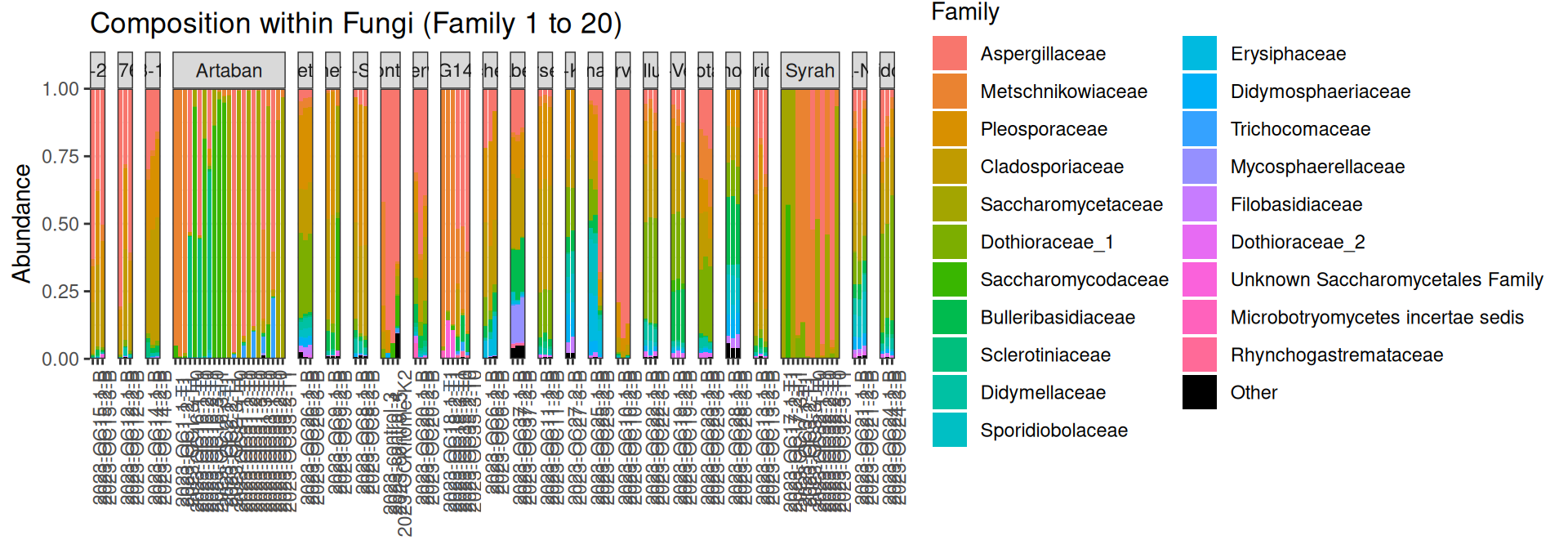
Here are the taxonomies after applying changes:
tib <- psmelt(physeq_updated) %>% as_tibble() %>% mutate(Taxo = paste(Kingdom,Phylum,Class,Order,Family,Genus,Species,sep=";")) %>% group_by(OTU) %>% ungroup() %>% select(OTU,Taxo) %>% unique()
datatable(tib)# tib_oenovardocc2 <- psmelt(physeq) %>% as_tibble() %>% mutate(Taxo = paste(Kingdom,Phylum,Class,Order,Family,Genus,Species,sep=";")) %>% group_by(OTU) %>% ungroup() %>% select(OTU,Taxo) %>% unique()
#
# tib_oenovardocc1 <- psmelt(oenovardocc1) %>% as_tibble() %>% mutate(Species = gsub(" ","_",Species)) %>% mutate(Taxo = paste(Kingdom,Phylum,Class,Order,Family,Genus,Species,sep=";")) %>% group_by(OTU) %>% ungroup() %>% select(OTU,Taxo) %>% unique()
tibjoined <- full_join(tibO2, tibO1, by="OTU") %>% mutate(Oenovardocc2 = Taxo.x, Oenovardocc1 = Taxo.y) %>% select(-Taxo.x, -Taxo.y)
tibupdated <- psmelt(physeq_updated) %>% as_tibble() %>% mutate(Taxo = paste(Kingdom,Phylum,Class,Order,Family,Genus,Species,sep=";")) %>% group_by(OTU) %>% ungroup() %>% select(OTU,Taxo) %>% unique()
tibfinal <- full_join(tibjoined, tibupdated, by="OTU")
write.table(tibfinal, "html/affis_final.txt", append=TRUE, quote = FALSE, row.names = FALSE, col.names = TRUE, sep = "\t" )
saveRDS(physeq_updated,"html/physeq_curated_with_oenovardocc1.rds")The changes can be viewed with Excel:
Merge of both runs
sample_data(oenovardocc1)$Year <- "2023"
sample_data(physeq_updated)$ID <- "O1"
#sample_names(oenovardocc1) <- glue::glue("{sample_names(oenovardocc1)}_O1")
#taxa_names(oenovardocc1) <- paste(taxa_names(oenovardocc1), "O1", sep = "_")
sample_data(physeq_updated)$Year <- "2023"
sample_data(physeq_updated)$ID <- "O2"
#sample_names(physeq_updated) <- glue::glue("{sample_names(physeq_updated)}_O2")
#taxa_names(physeq_updated) <- paste(taxa_names(physeq_updated), "02", sep = "_")
oenovardocc_merge <- merge_phyloseq(oenovardocc1, physeq_updated)
oenovardocc_mergephyloseq-class experiment-level object
otu_table() OTU Table: [ 619 taxa and 248 samples ]
sample_data() Sample Data: [ 248 samples by 12 sample variables ]
tax_table() Taxonomy Table: [ 619 taxa by 7 taxonomic ranks ]metadata_all <- read.table("data/Run1etRun2-2023_OenoVardOcc_Metadata.tsv", row.names = 1, header = TRUE, sep = "\t", stringsAsFactors = FALSE)
metadata_all <- metadata_all %>%
mutate(SampleName = rownames(metadata_all))
sample_data(oenovardocc_merge) <- metadata_all
saveRDS(oenovardocc_merge, file="html/oenovardocc_merge_brix.rds")microbiome::summarize_phyloseq(oenovardocc_merge)[[1]]
[1] "1] Min. number of reads = 0"
[[2]]
[1] "2] Max. number of reads = 183866"
[[3]]
[1] "3] Total number of reads = 20940165"
[[4]]
[1] "4] Average number of reads = 85820.3483606557"
[[5]]
[1] "5] Median number of reads = 86627.5"
[[6]]
[1] "7] Sparsity = 0.911133769432453"
[[7]]
[1] "6] Any OTU sum to 1 or less? NO"
[[8]]
[1] "8] Number of singletons = 0"
[[9]]
[1] "9] Percent of OTUs that are singletons \n (i.e. exactly one read detected across all samples)0"
[[10]]
[1] "10] Number of sample variables are: 12"
[[11]]
[1] "Type" "Variety_name" "Variety_type" "colour" "treatment"
[6] "locality" "Year" "inoculation" "collect.date" "modality"
[11] "Brix" "SampleName" We removed samples with less than 1000 reads.
ps <- prune_samples(sample_sums(oenovardocc_merge) > 10000, oenovardocc_merge)
psphyloseq-class experiment-level object
otu_table() OTU Table: [ 619 taxa and 238 samples ]
sample_data() Sample Data: [ 238 samples by 12 sample variables ]
tax_table() Taxonomy Table: [ 619 taxa by 7 taxonomic ranks ]microbiome::summarize_phyloseq(ps)[[1]]
[1] "1] Min. number of reads = 19563"
[[2]]
[1] "2] Max. number of reads = 183866"
[[3]]
[1] "3] Total number of reads = 20937535"
[[4]]
[1] "4] Average number of reads = 87972.8361344538"
[[5]]
[1] "5] Median number of reads = 87919.5"
[[6]]
[1] "7] Sparsity = 0.909226049062598"
[[7]]
[1] "6] Any OTU sum to 1 or less? NO"
[[8]]
[1] "8] Number of singletons = 0"
[[9]]
[1] "9] Percent of OTUs that are singletons \n (i.e. exactly one read detected across all samples)0"
[[10]]
[1] "10] Number of sample variables are: 12"
[[11]]
[1] "Type" "Variety_name" "Variety_type" "colour" "treatment"
[6] "locality" "Year" "inoculation" "collect.date" "modality"
[11] "Brix" "SampleName" saveRDS(ps, "html/physeq_curated_with_oenovardocc1_pruned.rds")Affiliations curation
Cécile and Gabriella modified some affiliations.
First, some of the ASVs have to be removed
#taxa_names(ps) <- gsub("_2023$", "", taxa_names(ps))
#taxa_names(ps) <- gsub("_2024$", "", taxa_names(ps))
correction_table <- read.csv2("html/affis_final_corrected.txt", sep = "\t", colClasses = c("character","character","character","character","character","character")) %>% as_tibble()
to_remove <- correction_table %>% filter(Taxo_Cecile == "a supprimer") %>% select(OTU) %>% pull()
to_remove <- paste0(to_remove,"_2024")
keptTaxa <- setdiff(taxa_names(ps), to_remove)
ps2 <- prune_taxa(keptTaxa, ps)And some have to be changed:
change_complete_taxo <- function(t, taxo, sequence){
taxolist <- unlist(strsplit(taxo, ";"))
sequence1 <- paste0(sequence,"_2023")
if(sequence1 %in% rownames(t)){
t[sequence1,"Kingdom"] <- taxolist[1]
t[sequence1,"Phylum"] <- taxolist[2]
t[sequence1,"Class"] <- taxolist[3]
t[sequence1,"Order"] <- taxolist[4]
t[sequence1,"Family"] <- taxolist[5]
t[sequence1,"Genus"] <- taxolist[6]
t[sequence1,"Species"] <- taxolist[7]
}
sequence2 <- paste0(sequence,"_2024")
if(sequence2 %in% rownames(t)){
t[sequence2,"Kingdom"] <- taxolist[1]
t[sequence2,"Phylum"] <- taxolist[2]
t[sequence2,"Class"] <- taxolist[3]
t[sequence2,"Order"] <- taxolist[4]
t[sequence2,"Family"] <- taxolist[5]
t[sequence2,"Genus"] <- taxolist[6]
t[sequence2,"Species"] <- taxolist[7]
}
return(t)
}
tib_changes_to_do <- correction_table %>% filter(Taxo_Cecile != "") %>% filter(Taxo_Cecile != "a supprimer") %>% select(OTU,Taxo_Cecile)
t <- as.data.frame(tib_changes_to_do)
tt <- phyloseq::tax_table(ps2)
for (i in 1:nrow(t)) {
tt <- change_complete_taxo(tt, t$Taxo_Cecile[i], t$OTU[i])
}
phyloseq::tax_table(ps2) <- tt
saveRDS(ps2, "html/final_physeq_brix.rds")Here are the final affiliations:
tib <- psmelt(ps2) %>% as_tibble() %>% mutate(Taxo = paste(Kingdom,Phylum,Class,Order,Family,Genus,Species,sep=";")) %>% group_by(OTU) %>% ungroup() %>% select(OTU,Taxo) %>% unique()
datatable(tib)Downloads
References
1. Shen W, Le S, Li Y, Hu F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/q file manipulation. PloS one. 2016;11:e0163962.
2. Andrews S. FastQC a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
3. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–8.
4. Escudié F, Auer L, Bernard M, Mariadassou M, Cauquil L, Vidal K, et al. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics. 2018;34:1287–94. doi:10.1093/bioinformatics/btx791.
5. Bernard M, Rué O, Mariadassou M, Pascal G. FROGS: a powerful tool to analyse the diversity of fungi with special management of internal transcribed spacers. Briefings in Bioinformatics. 2021;22. doi:10.1093/bib/bbab318.
6. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from illumina amplicon data. Nature methods. 2016;13:581.
7. Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: A fast and accurate illumina paired-end reAd mergeR. Bioinformatics. 2013;30:614–20.
8. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet journal. 2011;17:10–2.
Reuse
This document will not be accessible without prior agreement of the partners
