
cd /home/orue/work/PROJECTS/MARCO-BOLO/AQUARIUM_12S
unzip Raw_fastq-20240918T113509Z-001.zip
cd Raw_fastq/
for i in *.fastq.gz ; do id=$(echo $i |cut -d '_' -f 1); ext=$(echo $i |cut -d '_' -f 2); mv $i ${id}_R${ext} ; done
cd ../
mv Raw_fastq RAW_DATA
gzip -d Reference_library_12S_AllFish_Dez2023__LineageInfo_sintax_format.fasta.gz
grep -c '>' Reference_library_12S_AllFish_Dez2023__LineageInfo_sintax_format.fasta
32461
## Index FASTA
conda activate frogs-5.0.0
makeblastdb -in Reference_library_12S_AllFish_Dez2023__LineageInfo_sintax_formatFROGS.fasta -dbtype nucl
conda deactivate
## Make the archive
cd RAW_DATA
tar zcvf 12S.tar.gz *.fastq.gz
cd ../
Note
This document is a report of the analyses performed. You will find all the code used to analyze these data. The version of the tools (maybe in code chunks) and their references are indicated, for questions of reproducibility.
The MARCO-BOLO DATA ANALYSIS CHALLENGE
Join the EU Horizon MARCO-BOLO project in comparing bioinformatics pipelines for analysing eDNA data by participating in the DATA ANALYSIS CHALLENGE. In doing so, you will help improve recommendations for eDNA metabarcoding pipeline choice amongst the myriad of options out there and contribute towards the development of indicator workflows to report on biodiversity monitoring.
What you do
- 🔧 Come with your favourite pipeline
- 💻 Run it on one of our plankton or fish eDNA datasets
- 🌐 Send us your resulting OTU/ASV/ZOTU and Taxonomy tables
What we do
- 💾 Provide the eDNA datasets (18S plankton time series and 12S/16S/COI aquarium samples) along with standardised reference libraries
- 📊 Compile and compare the resulting tables across participants’ pipelines
- 📝 Report and share these results back with the participants and on the MARCO-BOLO website
Raw data
Data were provided by organizers through web links:
The files are demultiplexed and compressed to the “fastq.gz” format.
All the sequence associated files (sample metadata, primer information and template for registering pipeline information) are in the github page.
The primers are:
Forward primers
>milobo_COI_FWD
GGWACWGGWTGAACWGTWTAYCCYCC
>MiFish-U_12S_FWD
GTCGGTAAAACTCGTGCCAGC
>MiFish-E_12S_FWD
GTTGGTAAATCTCGTGCCAGC
>Fish16_16S_FWD
GACCCTATGGAGCTTTAGAC
Reverse primers
>milobo_COI_REV
TAAACYTCWGGRTGWCCRAARAAYCA
>MiFish-U_12S_REV
CATAGTGGGGTATCTAATCCCAGTTTG
>MiFish-E_12S_REV
CATAGTGGGGTATCTAATCCTAGTTTG
>Fish16_16S_REV
CGCTGTTATCCCTADRGTAACT
Warning
2 primers have been used for amplification. For forward primers, 2 positions are ambigous. We apply a IUPAC code: >MiFish-U_12S_FWD GTCGGTAAAACTCGTGCCAGC >MiFish-E_12S_FWD GTTGGTAAATCTCGTGCCAGC
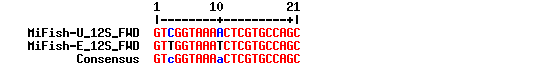
gives GTYGGTAAAWCTCGTGCCAGC
For reverse primers:
MiFish-U_12S_REV CATAGTGGGGTATCTAATCCCAGTTTG MiFish-E_12S_REV CATAGTGGGGTATCTAATCCTAGTTTG

gives CATAGTGGGGTATCTAATCCYAGTTTG
Data preparation
Data preparation means formatting all files in the right format for analysis with FROGS:
- Put all FASTQ files, renamed when needed, in a compressed archive
- Format reference FASTA files with Blast
[1] (available with FROGS [2] tool) for 12S, 16S and COI datasets - For the 18S dataset, a re-orientation of the reads is needed. It can be done with cutadapt
[3]
cd ~/work/PROJECTS/MARCO-BOLO/AQUARIUM_FISH16S/
unzip Raw_fastq-20240912T131951Z-001.zip
cd Raw_fastq/
for i in *.fastq.gz ; do id=$(echo $i |cut -d '_' -f 1); ext=$(echo $i |cut -d '_' -f 2); mv $i ${id}_R${ext} ; done
cd ../
mv Raw_fastq RAW_DATA
gzip -d Reference_library_16S_AllFish_Dez2023_LineageInfo_sintax_format.fasta.gz
grep -c '>' Reference_library_16S_AllFish_Dez2023_LineageInfo_sintax_format.fasta
37825
sed -e "s/,/;/g" -e "s/;tax=/\t/g" Reference_library_16S_AllFish_Dez2023_LineageInfo_sintax_format.fasta > Reference_library_16S_AllFish_Dez2023_LineageInfo_sintax_formatFROGS.fasta
## Index FASTA
conda activate frogs-5.0.0
makeblastdb -in Reference_library_16S_AllFish_Dez2023_LineageInfo_sintax_formatFROGS.fasta -dbtype nucl
conda deactivate
## Make the archive
cd RAW_DATA
tar zcvf 16S.tar.gz *.fastq.gz
cd ../unzip Raw_fastq-20240912T131951Z-001.zip
cd Raw_fastq/
for i in *.fastq.gz ; do id=$(echo $i |cut -d '_' -f 1); ext=$(echo $i |cut -d '_' -f 2); mv $i ${id}_R${ext} ; done
cd ../
mv Raw_fastq RAW_DATA
gzip -d Reference_library_COI_Metazoa_Sep2024_sintax_worms_tax.fasta.gz
grep -c '>' Reference_library_COI_Metazoa_Sep2024_sintax_worms_tax.fasta
390427
sed -e "s/,/;/g" -e "s/;tax=/\t/g" Reference_library_COI_Metazoa_Sep2024_sintax_worms_tax.fasta | perl -F'\t' -lane 'print "$F[0]$.\t$F[1]"' | sed 's/ /_/g' - > Reference_library_COI_Metazoa_Sep2024_sintax_worms_tax_formatFROGS.fasta
## Index FASTA
conda activate frogs-5.0.0
makeblastdb -in Reference_library_COI_Metazoa_Sep2024_sintax_worms_tax_formatFROGS.fasta -dbtype nucl
conda deactivate
## Make the archive
cd RAW_DATA
tar zcvf AquariumCOI.tar.gz *.fastq.gz
cd ../cd /home/orue/work/PROJECTS/MARCO-BOLO/PLANKTON_18S
unzip 3um_samples_fastq_files.tar.gz
cd Raw_fastq
for i in *.fastq.gz ; do id=$(echo $i |cut -d '_' -f 1); ext=$(echo $i |cut -d '_' -f 3); mv $i ${id}_${ext} ; done
cd ../
mv 3um_samples_fastq_files RAW_DATA
conda activate cutadapt-4.9
mkdir CUTADAPT
for i in RAW_DATA/*_R1.fastq.gz ; do id=$(echo $(basename $i) |cut -d '_' -f 1) ; cutadapt -g CCAGCASCYGCGGTAATTCC -G ACTTTCGTTCTTGATYRA --match-read-wildcard $i RAW_DATA/${id}_R2.fastq.gz -o CUTADAPT/${id}_R1.fastq.gz -p CUTADAPT/${id}_R2.fastq.gz --action none --revcomp ; done
conda deactivate
## Make the archive
cd CUTADAPT
tar zcvf 18S.tar.gz *.fastq.gz
cd ../Quality control
The quality control performed here allows to check the quality of the data:
- number of reads
- length of reads
- base qualities
conda activate seqkit-2.0.0 && seqkit stat RAW_DATA/*.fastq.gz > seqkit.txt && conda deactivate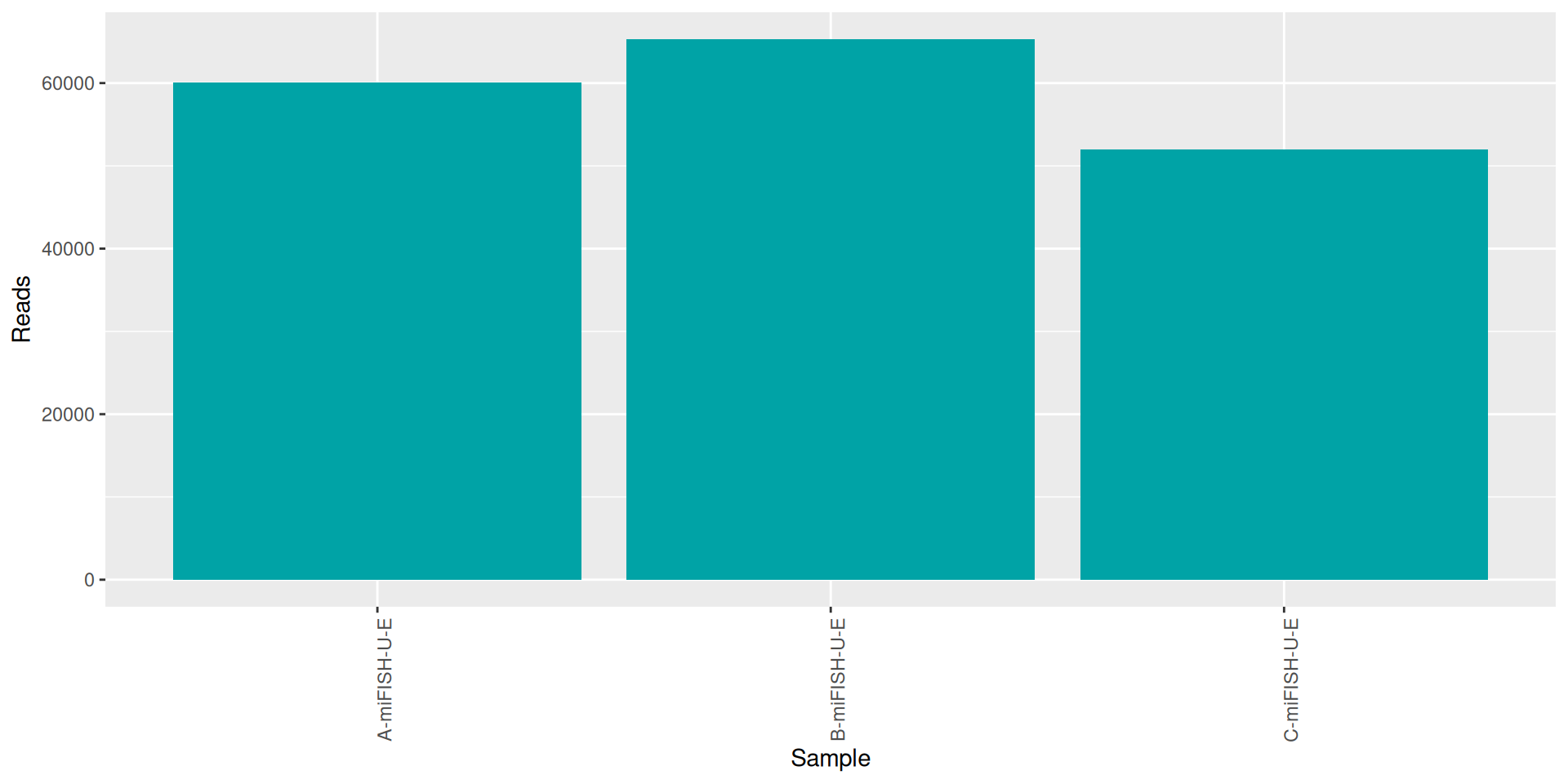
| sample | num_seqs | sum_len | min_len | avg_len | max_len |
|---|---|---|---|---|---|
| A-miFISH-U-E | 60052 | 17,185,292 | 35 | 286.2 | 301 |
| B-miFISH-U-E | 65329 | 18,681,448 | 35 | 286.0 | 301 |
| C-miFISH-U-E | 51947 | 14,762,596 | 35 | 284.2 | 301 |
mkdir FASTQC LOGS
for i in RAW_DATA/*.gz ; do echo "conda activate fastqc-0.11.9 && fastqc -t 8 $i -o FASTQC && conda deactivate" >> fastqc.sh ; done
qarray -cwd -V -N fastqc -pe thread 8 -o LOGS -e LOGS fastqc.sh
conda activate multiqc-1.11 && multiqc FASTQC -o MULTIQC && conda deactivateThe MultiQC report shows expected metrics for Illumina Miseq sequencing data.
conda activate seqkit-2.0.0 && seqkit stat RAW_DATA/*.fastq.gz > seqkit.txt && conda deactivate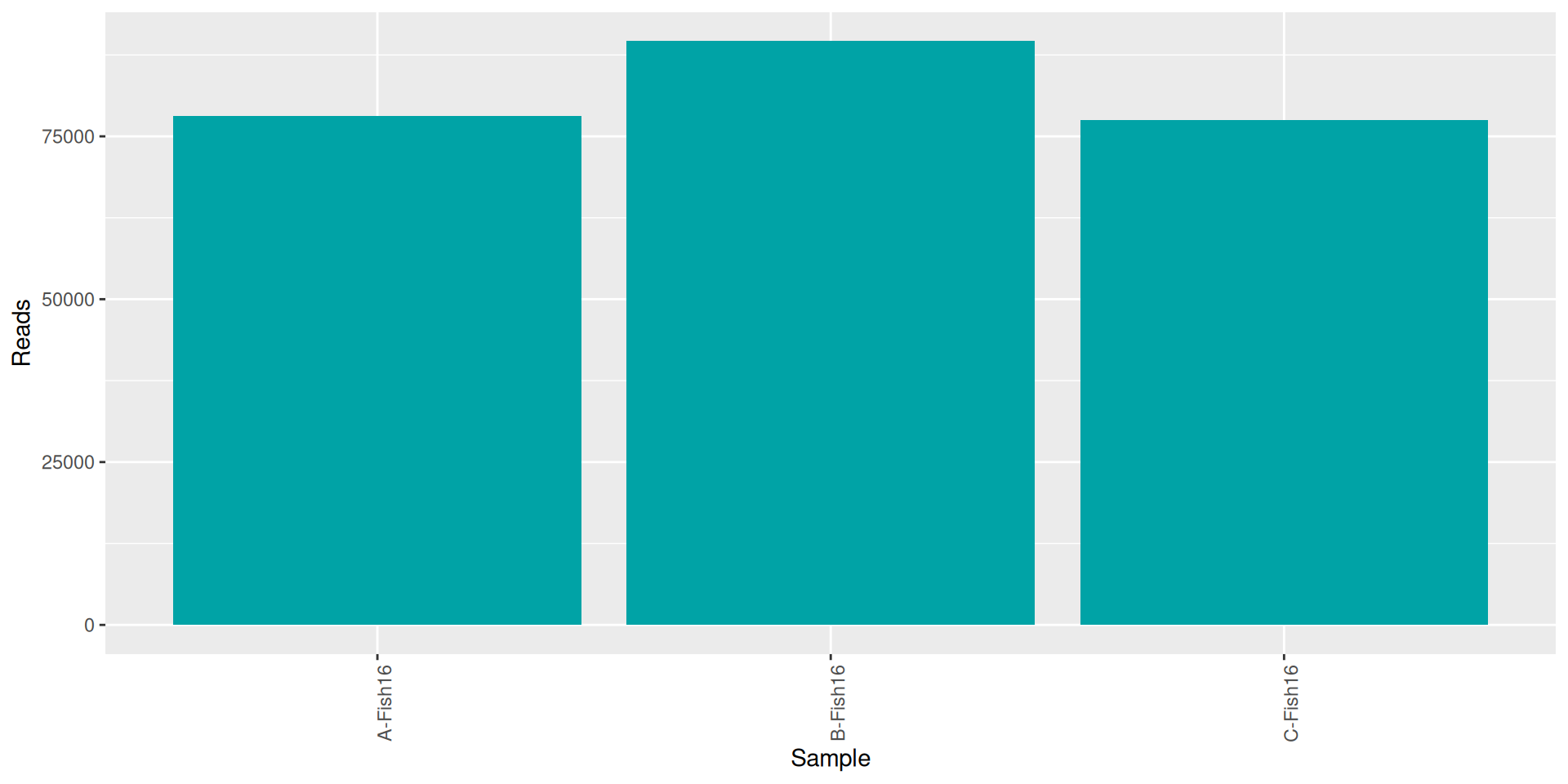
| sample | num_seqs | sum_len | min_len | avg_len | max_len |
|---|---|---|---|---|---|
| A-Fish16 | 78121 | 19,537,026 | 35 | 250.1 | 301 |
| B-Fish16 | 89623 | 21,535,751 | 35 | 240.3 | 301 |
| C-Fish16 | 77568 | 19,029,807 | 35 | 245.3 | 301 |
mkdir FASTQC LOGS
for i in RAW_DATA/*.gz ; do echo "conda activate fastqc-0.11.9 && fastqc -t 8 $i -o FASTQC && conda deactivate" >> fastqc.sh ; done
qarray -cwd -V -N fastqc -pe thread 8 -o LOGS -e LOGS fastqc.sh
conda activate multiqc-1.11 && multiqc FASTQC -o MULTIQC && conda deactivateThe MultiQC report shows expected metrics for Illumina Miseq sequencing data.
conda activate seqkit-2.0.0 && seqkit stat RAW_DATA/*.fastq.gz > seqkit.txt && conda deactivate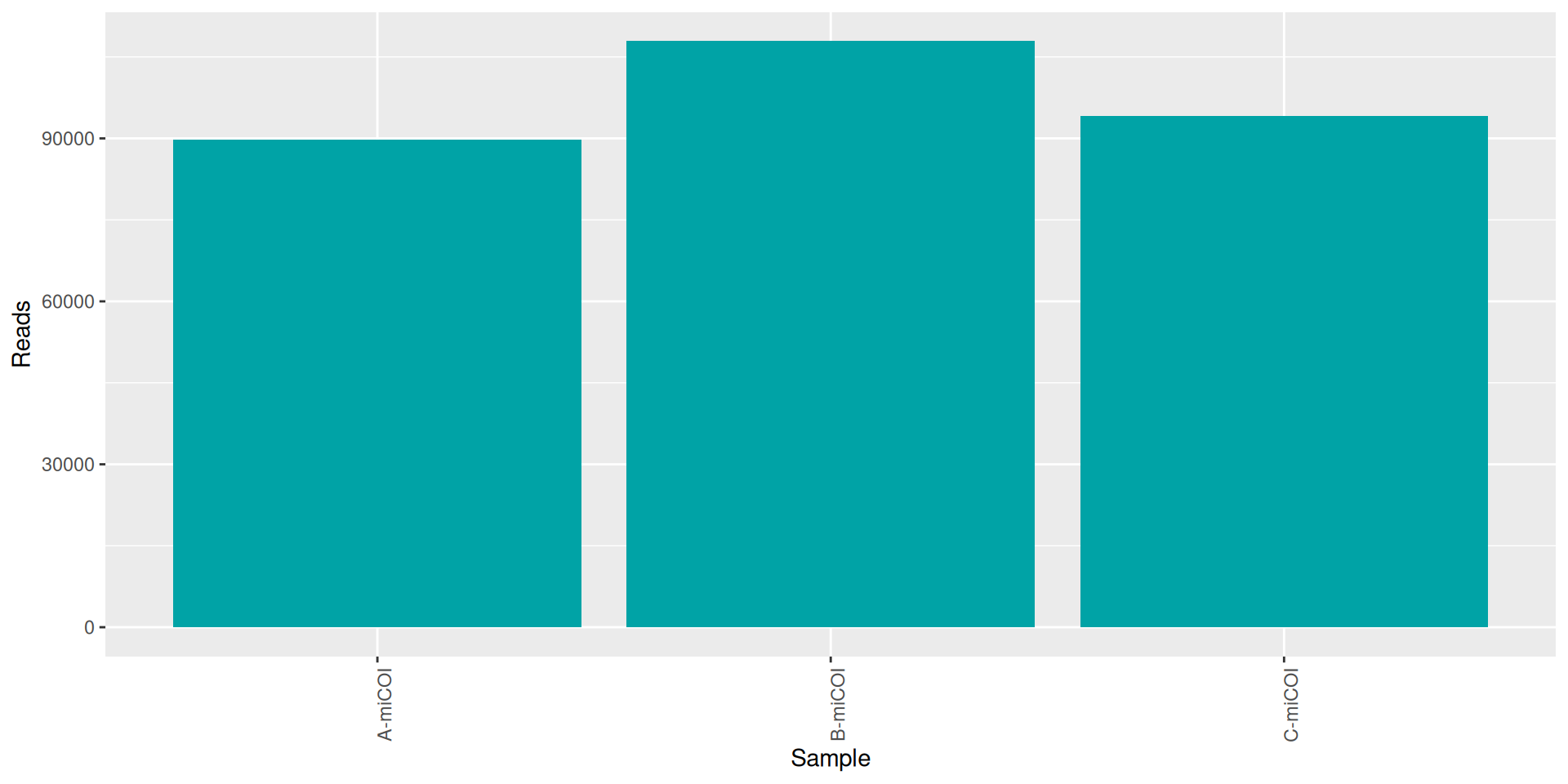
| sample | num_seqs | sum_len | min_len | avg_len | max_len |
|---|---|---|---|---|---|
| A-miCOI | 89833 | 26,339,388 | 35 | 293.2 | 301 |
| B-miCOI | 107889 | 31,518,638 | 35 | 292.1 | 301 |
| C-miCOI | 94108 | 27,578,552 | 35 | 293.1 | 301 |
mkdir FASTQC LOGS
for i in RAW_DATA/*.gz ; do echo "conda activate fastqc-0.11.9 && fastqc -t 8 $i -o FASTQC && conda deactivate" >> fastqc.sh ; done
qarray -cwd -V -N fastqc -pe thread 8 -o LOGS -e LOGS fastqc.sh
conda activate multiqc-1.11 && multiqc FASTQC -o MULTIQC && conda deactivateThe MultiQC report shows expected metrics for Illumina Miseq sequencing data.
conda activate seqkit-2.0.0 && seqkit stat RAW_DATA/*.fastq.gz > seqkit.txt && conda deactivate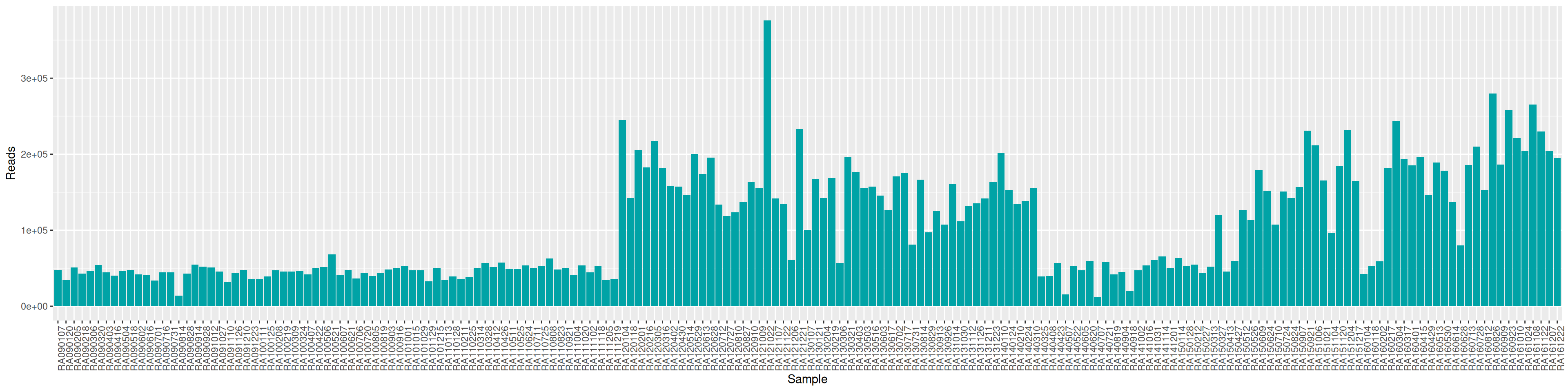
mkdir FASTQC LOGS
for i in RAW_DATA/*.gz ; do echo "conda activate fastqc-0.11.9 && fastqc -t 8 $i -o FASTQC && conda deactivate" >> fastqc.sh ; done
qarray -cwd -V -N fastqc -pe thread 8 -o LOGS -e LOGS fastqc.sh
conda activate multiqc-1.11 && multiqc FASTQC -o MULTIQC && conda deactivateThe MultiQC report shows expected metrics for Illumina Miseq sequencing data.
Note
All datasets are ready to be analyzed with FROGS.
FROGS v5.0.0 using swarm clustering step
- The tool
denoising.pyis set to perform the following actions:
- merge denoised reads with PEAR
[8] - keep only extended sequences
- remove adapters with cutadapt
[3] - filter sequences (min: 55, max: 550)
- dereplicate sequences
- cluster reads with swarm
[9] - generate a BIOM file
The tool
remove_chimera.pyuses vsearch[10] to remove chimera with parameter --uchime_denovo. Then, a cross-validation is performed.The tool
cluster_filters.pyallows to filter clusters by looking for contamination with phiX sequences (alignment with BLAST[1]), by removing low-abundant ( --min-abundance) and low-prevalent (--min-sample-presence) clusters.The tool
taxonomic_affiliation.pyuses BLAST[1] to align ASV sequences against the formated databank previously indexed. If there are several best hits, a Multi-affiliation tag is applied to the rank where there is ambiguity. The tool
biom_to_tsv.pyallows to transform the BIOM file into two tabulated files, one containing the ASV identifier, the ASV sequence, the counts per sample and the alignment metrics, the other containing any ambiguities for each multi-affiliated ASV.
Important
The following commands have been launched on the Genotoul infrastructure.
cd /work/project/frogs/MARCO-BOLO
module load devel/Miniconda/Miniconda3
module load bioinfo/FROGS/FROGS-v5.0.0
module load bioinfo/PEAR/0.9.10
CURRENT=AQUARIUM_12S
mkdir $CURRENT/Results
RES_DIR=$CURRENT/Results
mkdir $CURRENT/Logs
LOG_DIR=$CURRENT/Logs
SEQ_DIR=$CURRENT/RAW_DATA
INPUT=$SEQ_DIR/12S.tar.gz
FIVE_PRIM=GTYGGTAAAWCTCGTGCCAGC
THREE_PRIM=CAAACTRGGATTAGATACCCCACTATG
MIN=55
MAX=550
R1_SIZE=300
R2_SIZE=300
DATABQ=$CURRENT/Reference_library_12S_AllFish_Dez2023__LineageInfo_sintax_formatFROGS.fasta
PHIX=/save/user/frogs/galaxy_databanks/phiX_genome/phi.fa
sbatch -t 20 -J ${CURRENT}_denoising -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --cpus-per-task=16 --mem=240G --wrap="denoising.py illumina --merge-software pear --min-amplicon-size $MIN --max-amplicon-size $MAX --five-prim-primer $FIVE_PRIM --three-prim-primer $THREE_PRIM --R1-size $R1_SIZE --R2-size $R2_SIZE --nb-cpus 16 --output-fasta $RES_DIR/01-clusters.fasta --output-biom $RES_DIR/01-clusters.biom --html $RES_DIR/01-denoising.html --log-file $LOG_DIR/01-denoising.log --process swarm --input-archive $SEQ_DIR/12S.tar.gz"
sbatch -t 20 -J ${CURRENT}_rm_chim -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --cpus-per-task=16 --mem=240G --wrap="remove_chimera.py --input-fasta $RES_DIR/01-clusters.fasta --input-biom $RES_DIR/01-clusters.biom --output-fasta $RES_DIR/02-remove_chimera.fasta --nb-cpus 16 --log-file $LOG_DIR/02-r
emove_chimera.log --output-biom $RES_DIR/02-remove_chimera.biom --html $RES_DIR/02-remove_chimera.html"
sbatch -t 20 -J ${CURRENT}_filter --cpus-per-task=16 --mem=64G -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --wrap="cluster_filters.py --input-fasta $RES_DIR/02-remove_chimera.fasta --input-biom $RES_DIR/02-remove_chimera.biom --output-fasta $RES_DIR/03-filters.fasta --nb-cpus 16 --log-file $LOG_DIR/
03-filters.log --output-biom $RES_DIR/03-filters.biom --html $RES_DIR/03-filters.html --excluded $RES_DIR/03-filters_excluded.tsv --contaminant $PHIX --min-sample-presence 2 --min-abundance 0.00005"
sbatch -t 20 -J ${CURRENT}_affi --cpus-per-task=16 --mem=240G -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --wrap="taxonomic_affiliation.py --input-fasta $RES_DIR/03-filters.fasta --input-biom $RES_DIR/03-filters.biom --nb-cpus 16 --log-file $LOG_DIR/04-affiliation.log --output-biom $RES_DIR/04-affil
iation.biom --html $RES_DIR/04-affiliation.html --reference $DATABQ"
sbatch -t 20 -J ${CURRENT}_biom_to_tsv -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --wrap="biom_to_tsv.py --input-biom $RES_DIR/04-affiliation.biom --output-tsv $RES_DIR/05-affiliation.tsv --output-multi-affi $RES_DIR/05-affiliation.multihit.tsv --input-fasta $RES_DIR/03-filters.fasta --log-file $LO
G_DIR/05-biom_to_tsv.log"
Note
- For the denoising step, we keep 87% of sequences. 20,270 clusters are generated, corresponding to 154,501 sequences.
- A low chimera rate is observed (10% of sequences lost: 9,907 clusters corresponding to 14,910 sequences).
- Filters (0.005% -> 7 sequences and presence in at least 2 samples) allow to remove 9,193 clusters and 10% of sequences (13,682).
- 51 / 1,170 ASVs are affiliated (10% of the sequences) against the provided databank.
module load devel/Miniconda/Miniconda3
module load bioinfo/FROGS/FROGS-v5.0.0
module load bioinfo/PEAR/0.9.10
CURRENT=AQUARIUM_FISH16S
mkdir $CURRENT/Results
RES_DIR=$CURRENT/Results
mkdir $CURRENT/Logs
LOG_DIR=$CURRENT/Logs
SEQ_DIR=$CURRENT/RAW_DATA
INPUT=$SEQ_DIR/16S.tar.gz
FIVE_PRIM=GACCCTATGGAGCTTTAGAC
THREE_PRIM=AGTTACYHTAGGGATAACAGCG
MIN=55
MAX=550
R1_SIZE=300
R2_SIZE=300
DATABQ=$CURRENT/Reference_library_16S_AllFish_Dez2023_LineageInfo_sintax_formatFROGS.fasta
PHIX=/save/user/frogs/galaxy_databanks/phiX_genome/phi.fa
sbatch -t 20 -J ${CURRENT}_denoising -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --cpus-per-task=16 --mem=240G --wrap="denoising.py illumina --merge-software pear --min-amplicon-size $MIN --max-amplicon-size $MAX --five-prim-primer $FIVE_PRIM --three-prim-primer $THREE_PRIM --R1-size $R1_SIZE --R2-size $R2_SIZE --nb-cpus 16 --output-fasta $RES_DIR/01-clusters.fasta --output-biom $RES_DIR/01-clusters.biom --html $RES_DIR/01-denoising.html --log-file $LOG_DIR/01-denoising.log --process swarm --input-archive $INPUT"
sbatch -t 20 -J ${CURRENT}_rm_chim -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --cpus-per-task=16 --mem=240G --wrap="remove_chimera.py --input-fasta $RES_DIR/01-clusters.fasta --input-biom $RES_DIR/01-clusters.biom --output-fasta $RES_DIR/02-remove_chimera.fasta --nb-cpus 16 --log-file $LOG_DIR/02-remove_chimera.log --output-biom $RES_DIR/02-remove_chimera.biom --html $RES_DIR/02-remove_chimera.html"
sbatch -t 20 -J ${CURRENT}_filter --cpus-per-task=16 --mem=64G -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --wrap="cluster_filters.py --input-fasta $RES_DIR/02-remove_chimera.fasta --input-biom $RES_DIR/02-remove_chimera.biom --output-fasta $RES_DIR/03-filters.fasta --nb-cpus 16 --log-file $LOG_DIR/03-filters.log --output-biom $RES_DIR/03-filters.biom --html $RES_DIR/03-filters.html --excluded $RES_DIR/03-filters_excluded.tsv --contaminant $PHIX --min-sample-presence 2 --min-abundance 0.00005"
sbatch -t 20 -J ${CURRENT}_affi --cpus-per-task=16 --mem=240G -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --wrap="taxonomic_affiliation.py --input-fasta $RES_DIR/03-filters.fasta --input-biom $RES_DIR/03-filters.biom --nb-cpus 16 --log-file $LOG_DIR/04-affiliation.log --output-biom $RES_DIR/04-affiliation.biom --html $RES_DIR/04-affiliation.html --reference $DATABQ"
sbatch -t 20 -J ${CURRENT}_biom_to_tsv -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --wrap="biom_to_tsv.py --input-biom $RES_DIR/04-affiliation.biom --output-tsv $RES_DIR/05-affiliation.tsv --output-multi-affi $RES_DIR/05-affiliation.multihit.tsv --input-fasta $RES_DIR/03-filters.fasta --log-file $LOG_DIR/05-biom_to_tsv.log"
Note
- For the denoising step, we keep 75% of sequences. 20,402 clusters are generated, corresponding to 183,320 sequences.
- A low chimera rate is observed (6% of sequences lost: 8,390 clusters corresponding to 12,007 sequences).
- Filters (0.005% -> 9 sequences and presence in at least 2 samples) allow to remove 11,373 clusters and 8% of sequences (14,257).
- 40 / 639 ASVs are affiliated (5% of the sequences) against the provided databank.
module load devel/Miniconda/Miniconda3
module load bioinfo/FROGS/FROGS-v5.0.0
module load bioinfo/PEAR/0.9.10
CURRENT=AQUARIUM_COI
mkdir $CURRENT/Results
RES_DIR=$CURRENT/Results
mkdir $CURRENT/Logs
LOG_DIR=$CURRENT/Logs
SEQ_DIR=$CURRENT/RAW_DATA
INPUT=$SEQ_DIR/AquariumCOI.tar.gz
FIVE_PRIM=GGWACWGGWTGAACWGTWTAYCCYCC
THREE_PRIM=TGRTTYTTYGGWCAYCCWGARGTTTA
MIN=55
MAX=550
R1_SIZE=300
R2_SIZE=300
DATABQ=$CURRENT/Reference_library_COI_Metazoa_Sep2024_sintax_worms_tax_formatFROGS.fasta
PHIX=/save/user/frogs/galaxy_databanks/phiX_genome/phi.fa
sbatch -t 20 -J ${CURRENT}_denoising -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --cpus-per-task=16 --mem=240G --wrap="denoising.py illumina --merge-software pear --min-amplicon-size $MIN --max-amplicon-size $MAX --five-prim-primer $FIVE_PRIM --three-prim-primer $THREE_PRIM --R1-size $R1_SIZE --R2-size $R2_SIZE --nb-cpus 16 --output-fasta $RES_DIR/01-clusters.fasta --output-biom $RES_DIR/01-clusters.biom --html $RES_DIR/01-denoising.html --log-file $LOG_DIR/01-denoising.log --process swarm --input-archive $INPUT"
sbatch -t 20 -J ${CURRENT}_rm_chim -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --cpus-per-task=16 --mem=240G --wrap="remove_chimera.py --input-fasta $RES_DIR/01-clusters.fasta --input-biom $RES_DIR/01-clusters.biom --output-fasta $RES_DIR/02-remove_chimera.fasta --nb-cpus 16 --log-file $LOG_DIR/02-remove_chimera.log --output-biom $RES_DIR/02-remove_chimera.biom --html $RES_DIR/02-remove_chimera.html"
sbatch -t 20 -J ${CURRENT}_filter --cpus-per-task=16 --mem=64G -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --wrap="cluster_filters.py --input-fasta $RES_DIR/02-remove_chimera.fasta --input-biom $RES_DIR/02-remove_chimera.biom --output-fasta $RES_DIR/03-filters.fasta --nb-cpus 16 --log-file $LOG_DIR/03-filters.log --output-biom $RES_DIR/03-filters.biom --html $RES_DIR/03-filters.html --excluded $RES_DIR/03-filters_excluded.tsv --contaminant $PHIX --min-sample-presence 2 --min-abundance 0.00005"
sbatch -t 20 -J ${CURRENT}_affi --cpus-per-task=16 --mem=240G -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --wrap="taxonomic_affiliation.py --input-fasta $RES_DIR/03-filters.fasta --input-biom $RES_DIR/03-filters.biom --nb-cpus 16 --log-file $LOG_DIR/04-affiliation.log --output-biom $RES_DIR/04-affiliation.biom --html $RES_DIR/04-affiliation.html --reference $DATABQ"
sbatch -t 20 -J ${CURRENT}_biom_to_tsv -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --wrap="biom_to_tsv.py --input-biom $RES_DIR/04-affiliation.biom --output-tsv $RES_DIR/05-affiliation.tsv --output-multi-affi $RES_DIR/05-affiliation.multihit.tsv --input-fasta $RES_DIR/03-filters.fasta --log-file $LOG_DIR/05-biom_to_tsv.log"
Note
- For the denoising step, we keep 85% of sequences. 13,205 clusters are generated, corresponding to 249,143 sequences.
- A very low chimera rate is observed (1% of sequences lost: 2,256 clusters corresponding to 3,088 sequences).
- Filters (0.005% -> 13 sequences and presence in at least 2 samples) allow to remove 10,268 clusters and 10% of sequences (25,488).
- 399 / 681 ASVs are affiliated (53% of the sequences) against the provided databank.
module load devel/Miniconda/Miniconda3
module load bioinfo/FROGS/FROGS-v5.0.0
module load bioinfo/PEAR/0.9.10
CURRENT=PLANKTON_18S
mkdir $CURRENT/Results
RES_DIR=$CURRENT/Results
mkdir $CURRENT/Logs
LOG_DIR=$CURRENT/Logs
SEQ_DIR=$CURRENT/RAW_DATA
INPUT=$SEQ_DIR/18S_well_oriented.tar.gz
FIVE_PRIM=CCAGCASCYGCGGTAATTCC
THREE_PRIM=TYRATCAAGAACGAAAGT
MIN=55
MAX=550
R1_SIZE=300
R2_SIZE=300
DATABQ=/save/user/frogs/galaxy_databanks/18S/PR2_5.0.1/PR2_5.0.1.fasta
PHIX=/save/user/frogs/galaxy_databanks/phiX_genome/phi.fa
sbatch -t 240 -J ${CURRENT}_denoising -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --cpus-per-task=16 --mem=240G --wrap="denoising.py illumina --merge-software pear --min-amplicon-size $MIN --max-amplicon-size $MAX --five-prim-primer $FIVE_PRIM --three-prim-primer $THREE_PRIM --R1-size $R1_SIZE --R2-size $R2_SIZE --nb-cpus 16 --output-fasta $RES_DIR/01-clusters.fasta --output-biom $RES_DIR/01-clusters.biom --html $RES_DIR/01-denoising.html --log-file $LOG_DIR/01-denoising.log --process swarm --input-archive $INPUT"
sbatch -t 20 -J ${CURRENT}_rm_chim -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --cpus-per-task=16 --mem=240G --wrap="remove_chimera.py --input-fasta $RES_DIR/01-clusters.fasta --input-biom $RES_DIR/01-clusters.biom --output-fasta $RES_DIR/02-remove_chimera.fasta --nb-cpus 16 --log-file $LOG_DIR/02-remove_chimera.log --output-biom $RES_DIR/02-remove_chimera.biom --html $RES_DIR/02-remove_chimera.html"
sbatch -t 20 -J ${CURRENT}_filter --cpus-per-task=16 --mem=64G -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --wrap="cluster_filters.py --input-fasta $RES_DIR/02-remove_chimera.fasta --input-biom $RES_DIR/02-remove_chimera.biom --output-fasta $RES_DIR/03-filters.fasta --nb-cpus 16 --log-file $LOG_DIR/03-filters.log --output-biom $RES_DIR/03-filters.biom --html $RES_DIR/03-filters.html --excluded $RES_DIR/03-filters_excluded.tsv --contaminant $PHIX --min-sample-presence 1 --min-abundance 0.00005"
sbatch -t 20 -J ${CURRENT}_affi --cpus-per-task=16 --mem=240G -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --wrap="taxonomic_affiliation.py --input-fasta $RES_DIR/03-filters.fasta --input-biom $RES_DIR/03-filters.biom --nb-cpus 16 --log-file $LOG_DIR/04-affiliation.log --output-biom $RES_DIR/04-affiliation.biom --html $RES_DIR/04-affiliation.html --reference $DATABQ --taxonomy-ranks Domain Supergroup Division Subdivision Class Order Family Genus Species"
sbatch -t 20 -J ${CURRENT}_biom_to_tsv -o $LOG_DIR/%x.out -e $LOG_DIR/%x.err --wrap="biom_to_tsv.py --input-biom $RES_DIR/04-affiliation.biom --output-tsv $RES_DIR/05-affiliation.tsv --output-multi-affi $RES_DIR/05-affiliation.multihit.tsv --input-fasta $RES_DIR/03-filters.fasta --log-file $LOG_DIR/05-biom_to_tsv.log"
Note
- For the denoising step, we keep 89% of sequences. 2,485,292 clusters are generated, corresponding to 17,364,580 sequences.
- A very low chimera rate is observed (1% of sequences lost: 142,456 clusters corresponding to 226,726 sequences).
- Filters (0.005% -> 857 sequences and presence in at least 1 sample) allow to remove 2,341,832 clusters and 19% of sequences (3,199,948).
- 1,002 / 1,004 ASVs are affiliated (99% of the sequences) against the PR2 v5.0.1 databank.
FROGS v5.0.0 using dada2 denoising step
- The tool
denoising.pyis set to perform the following actions:
- remove adapters with cutadapt
[3] - denoise reads with DADA2
[11] core algorithm - merge denoised reads with PEAR
[8] - keep only extended sequences
- filter sequences (min: 55, max: 550)
- dereplicate sequences
- generate a BIOM file
The tool
remove_chimera.pyuses vsearch[10] to remove chimera with parameter --uchime_denovo. Then, a cross-validation is performed.The tool
cluster_filters.pyallows to filter clusters by looking for contamination with phiX sequences (alignment with BLAST[1]), by removing low-abundant ( --min-abundance) and low-prevalent (--min-sample-presence) clusters.The tool
taxonomic_affiliation.pyuses BLAST[1] to align ASV sequences against the formated databank previously indexed. If there are several best hits, a Multi-affiliation tag is applied to the rank where there is ambiguity. The tool
biom_to_tsv.pyallows to transform the BIOM file into two tabulated files, one containing the ASV identifier, the ASV sequence, the counts per sample and the alignment metrics, the other containing any ambiguities for each multi-affiliated ASV.
Important
The following commands have been launched on the Migale infrastructure.
cd ~/work/PROJECTS/MARCO-BOLO/AQUARIUM_12S
conda activate frogs-5.0.0
denoising.py illumina --min-amplicon-size 55 --max-amplicon-size 550 --five-prim-primer GTYGGTAAAWCTCGTGCCAGC --three-prim-primer CAAACTRGGATTAGATACCCCACTATG --R1-size 300 --R2-size 300 --nb-cpus 16 --output-fasta clusters.fasta --output-biom clusters.biom --html denoising.html --log-file denoising.log --process dada2 --input-archive RAW_DATA/12S.tar.gz --sample-inference pseudo-pooling --merge-software pear
remove_chimera.py --input-fasta clusters.fasta --input-biom clusters.biom --output-fasta remove_chimera.fasta --nb-cpus 8 --log-file remove_chimera.log --output-biom remove_chimera.biom --html remove_chimera.html
cluster_filters.py --input-fasta remove_chimera.fasta --input-biom remove_chimera.biom --output-fasta filters.fasta --nb-cpus 8 --log-file filters.log --output-biom filters.biom --html filters.html --excluded filters_excluded.tsv --contaminant /db/outils/FROGS/contaminants/phi.fa --min-sample-presence 2 --min-abundance 0.00005
taxonomic_affiliation.py --input-fasta filters.fasta --input-biom filters.biom --nb-cpus 8 --log-file affiliation.log --output-biom affiliation.biom --html affiliation.html --reference Reference_library_12S_AllFish_Dez2023__LineageInfo_sintax_formatFROGS.fasta
biom_to_tsv.py --input-biom affiliation.biom --output-tsv affiliation.tsv --input-fasta filters.fasta --output-multi-affi multihits.tsv
Note
- For the denoising step, we keep 73% of sequences. 1,363 clusters are generated, corresponding to 129,696 sequences.
- A very low chimera rate is observed (1% of sequences lost: 129 clusters corresponding to 1,721 sequences).
- Filters (0.005% -> 7 sequences and presence in at least 2 samples) allow to remove 373 clusters and 6% of sequences (7,737).
- 38 / 373 ASVs are affiliated (10% of the sequences) against the provided databank.
cd ~/work/PROJECTS/MARCO-BOLO/AQUARIUM_FISH16S
conda activate frogs-5.0.0
denoising.py illumina --min-amplicon-size 55 --max-amplicon-size 550 --five-prim-primer GACCCTATGGAGCTTTAGAC --three-prim-primer AGTTACYHTAGGGATAACAGCG --R1-size 300 --R2-size 300 --nb-cpus 16 --output-fasta clusters.fasta --output-biom clusters.biom --html denoising.html --log-file denoising.log --process dada2 --input-archive RAW_DATA/16S.tar.gz --sample-inference pseudo-pooling --merge-software pear
remove_chimera.py --input-fasta clusters.fasta --input-biom clusters.biom --output-fasta remove_chimera.fasta --nb-cpus 8 --log-file remove_chimera.log --output-biom remove_chimera.biom --html remove_chimera.html
cluster_filters.py --input-fasta remove_chimera.fasta --input-biom remove_chimera.biom --output-fasta filters.fasta --nb-cpus 8 --log-file filters.log --output-biom filters.biom --html filters.html --excluded filters_excluded.tsv --contaminant /db/outils/FROGS/contaminants/phi.fa --min-sample-presence 2 --min-abundance 0.00005
taxonomic_affiliation.py --input-fasta filters.fasta --input-biom filters.biom --nb-cpus 8 --log-file affiliation.log --output-biom affiliation.biom --html affiliation.html --reference Reference_library_16S_AllFish_Dez2023_LineageInfo_sintax_formatFROGS.fasta
biom_to_tsv.py --input-biom affiliation.biom --output-tsv affiliation.tsv --input-fasta filters.fasta --output-multi-affi multihits.tsv
Note
- For the denoising step, we keep 68% of sequences. 1,869 clusters are generated, corresponding to 168,436 sequences.
- A very low chimera rate is observed (2% of sequences lost: 761 clusters corresponding to 2,884 sequences).
- Filters (0.005% -> 9 sequences and presence in at least 2 samples) allow to remove 683 clusters and 2% of sequences (3,943).
- Only 34 / 425 ASVs are affiliated (5% of the sequences!) against the provided databank.
cd ~/work/PROJECTS/MARCO-BOLO/AQUARIUM_COI
conda activate frogs-5.0.0
denoising.py illumina --min-amplicon-size 55 --max-amplicon-size 550 --five-prim-primer GGWACWGGWTGAACWGTWTAYCCYCC --three-prim-primer TGRTTYTTYGGWCAYCCWGARGTTTA --R1-size 300 --R2-size 300 --nb-cpus 16 --output-fasta clusters.fasta --output-biom clusters.biom --html denoising.html --log-file denoising.log --process dada2 --input-archive RAW_DATA/COI.tar.gz --merge-software pear
remove_chimera.py --input-fasta clusters.fasta --input-biom clusters.biom --output-fasta remove_chimera.fasta --nb-cpus 8 --log-file remove_chimera.log --output-biom remove_chimera.biom --html remove_chimera.html
cluster_filters.py --input-fasta remove_chimera.fasta --input-biom remove_chimera.biom --output-fasta filters.fasta --nb-cpus 8 --log-file filters.log --output-biom filters.biom --html filters.html --excluded filters_excluded.tsv --contaminant /db/outils/FROGS/contaminants/phi.fa --min-sample-presence 2 --min-abundance 0.00005
taxonomic_affiliation.py --input-fasta filters.fasta --input-biom filters.biom --nb-cpus 8 --log-file affiliation.log --output-biom affiliation.biom --html affiliation.html --reference Reference_library_COI_Metazoa_Sep2024_sintax_worms_tax_formatFROGS.fasta
biom_to_tsv.py --input-biom affiliation.biom --output-tsv affiliation.tsv --input-fasta filters.fasta --output-multi-affi multihits.tsv
Note
- For the denoising step, we keep 82% of sequences. 1,385 clusters are generated, corresponding to 240,492 sequences.
- A very low chimera rate is observed (<1% of sequences lost: 66 clusters corresponding to 601 sequences).
- Filters (0.005% -> 12 sequences and presence in at least 2 samples) allow to remove 797 clusters and 8% of sequences (20,165).
- 330 / 522 ASVs are affiliated (53% of the sequences) against the provided databank.
cd ~/work/PROJECTS/MARCO-BOLO/PLANKTON_18S
conda activate frogs-5.0.0
denoising.py illumina --min-amplicon-size 55 --max-amplicon-size 550 --five-prim-primer CCAGCASCYGCGGTAATTCC --three-prim-primer TYRATCAAGAACGAAAGT --R1-size 300 --R2-size 300 --nb-cpus 16 --output-fasta clusters.fasta --output-biom clusters.biom --html denoising.html --log-file denoising.log --process dada2 --input-archive CUTADAPT/18S.tar.gz --sample-inference pseudo-pooling --merge-software pear
remove_chimera.py --input-fasta clusters.fasta --input-biom clusters.biom --output-fasta remove_chimera.fasta --nb-cpus 8 --log-file remove_chimera.log --output-biom remove_chimera.biom --html remove_chimera.html
cluster_filters.py --input-fasta remove_chimera.fasta --input-biom remove_chimera.biom --output-fasta filters.fasta --nb-cpus 8 --log-file filters.log --output-biom filters.biom --html filters.html --excluded filters_excluded.tsv --contaminant /db
/outils/FROGS/contaminants/phi.fa --min-sample-presence 1 --min-abundance 0.00005
taxonomic_affiliation.py --input-fasta filters.fasta --input-biom filters.biom --nb-cpus 8 --log-file affiliation.log --output-biom affiliation.biom --html affiliation.html --reference /db/outils/FROGS/assignation/PR2_5.0.1/PR2_5.0.1.fasta --taxonomy-ranks Domain Supergroup Division Subdivision Class Order Family Genus Species
biom_to_tsv.py --input-biom affiliation.biom --output-tsv affiliation.tsv --input-fasta filters.fasta --output-multi-affi multihits.tsv
Note
- For the denoising step, we keep 92% of sequences. 116,969 clusters are generated, corresponding to 17,867,100 sequences.
- A very low chimera rate is observed (1% of sequences lost: 75,855 clusters (65% !) corresponding to 179,901 sequences).
- Filters (0.005% -> 885 sequences and presence in at least 1 sample) allow to remove 39,782 clusters and 6% of sequences (1,128,797).
- 1,331 / 1,332 ASVs are affiliated (99% of the sequences) against the PR2 v5.0.1 databank.
Downloads
FROGS v5.0.0 using swarm clustering step
FROGS v5.0.0 using dada2 denoising step
References
1. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of molecular biology. 1990;215:403–10.
2. Escudié F, Auer L, Bernard M, Mariadassou M, Cauquil L, Vidal K, et al. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics. 2018;34:1287–94. doi:10.1093/bioinformatics/btx791.
3. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet journal. 2011;17:10–2.
4. Shen W, Le S, Li Y, Hu F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/q file manipulation. PloS one. 2016;11:e0163962.
5. Andrews S. FastQC a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
6. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–8.
7. Bernard M, Rué O, Mariadassou M, Pascal G. FROGS: a powerful tool to analyse the diversity of fungi with special management of internal transcribed spacers. Briefings in Bioinformatics. 2021;22. doi:10.1093/bib/bbab318.
8. Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: A fast and accurate illumina paired-end reAd mergeR. Bioinformatics. 2013;30:614–20.
9. Mahé F, Rognes T, Quince C, Vargas C de, Dunthorn M. Swarm v2: Highly-scalable and high-resolution amplicon clustering. PeerJ. 2015;3:e1420.
10. Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: A versatile open source tool for metagenomics. PeerJ. 2016;4:e2584.
11. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from illumina amplicon data. Nature methods. 2016;13:581.
Reuse
This document will not be accessible without prior agreement of the partners
