
cd /home/orue/work/PROJECTS/LTMCBW/
mkdir RAW_DATA
cp /home/bigey/work/tmp/* RAW_DATA/
cd RAW_DATA
mv *16S* ../16S/
cd ../16S/
# Rename FASTQ files for 16S
for i in *_R1_001.fastq.gz ; do id=$(echo $(basename $i) | cut -d '_' -f 1) ; cp $i ${id}_R1.fastq.gz ; done
for i in *_R2_001.fastq.gz ; do id=$(echo $(basename $i) | cut -d '_' -f 1) ; cp $i ${id}_R2.fastq.gz ; done
rm -f *_001.fastq.gz
for i in *.fastq.gz ; do echo $i ; done |wc -l
126
#tar zcvf 16S.tar.gz *.fastq.gz
# ITS
cd ../RAW_DATA
mv *.fastq.gz ../ITS/
cd ../ITS
for i in *_R1_001.fastq.gz ; do id=$(echo $(basename $i) | cut -d '_' -f 1) ;cp $i ${id}_R1.fastq.gz ; done
for i in *_R2_001.fastq.gz ; do id=$(echo $(basename $i) | cut -d '_' -f 1) ;cp $i ${id}_R2.fastq.gz ; done
rm -f Undetermined_*
rm -f *_001.fastq.gz
for i in *.fastq.gz ; do echo $i ; done |wc -l
118
tar zcvf ITS.tar.gz *.fastq.gzLTMCBW
Microbial diversity analysis of Lebanese traditional monastic concentrated boiled wine by metabarcoding of 16S and ITS regions
closed
collaboration
Note
This document is a report of the analyses performed. You will find all the code used to analyze these data. The version of the tools (maybe in code chunks) and their references are indicated, for questions of reproducibility.
Aim of the project
The aim of this project is to provide the results of the FROGS5 preprocess tool which is not yet available to the community.
Patners
- Olivier Rué - Migale/MaIAGE - INRAE
- Pamela Bechara - SPO - INRAE
Deliverables
Deliverables agreed at the preliminary meeting (Table 1).
| Definition | |
|---|---|
| 1 | HTML report |
| 2 | Archive containing data to be stored |
| 3 | BIOM, FASTA and HTML files out from FROGS5 preprocess |
| 4 | BIOM file after affiliation with METABARFOOD and UNITE sequences |
Data management
Important
All data is managed by the migale facility for the duration of the project. Once the project is over, the Migale facility does not keep your data. We will provide you with the raw data and associated metadata that will be deposited on public repositories before the results are used. We can guide you in the submission process. We will then decide which files to keep, knowing that this report will also be provided to you and that the analyses can be replayed if needed.
Raw data
Raw data were deposited on the front server and a copy was sent to the abaca server.
Here is the number of samples for each amplicon:
| Dataset | Samples |
|---|---|
| 16S | 63 |
| ITS | 59 |
Quality control
We can plot and display the number of reads to see if enough reads are present and if samples are homogeneous.
cd /home/orue/work/PROJECTS/LTMCBW/16S/
mkdir LOGS
qsub -cwd -V -e LOGS -o LOGS -N seqkit -pe thread 4 -R y -b y "conda activate seqkit-2.0.0 && seqkit stats /home/orue/work/PROJECTS/LTMCBW/16S/*.fastq.gz -j 4 > raw_data.infos && conda deactivate"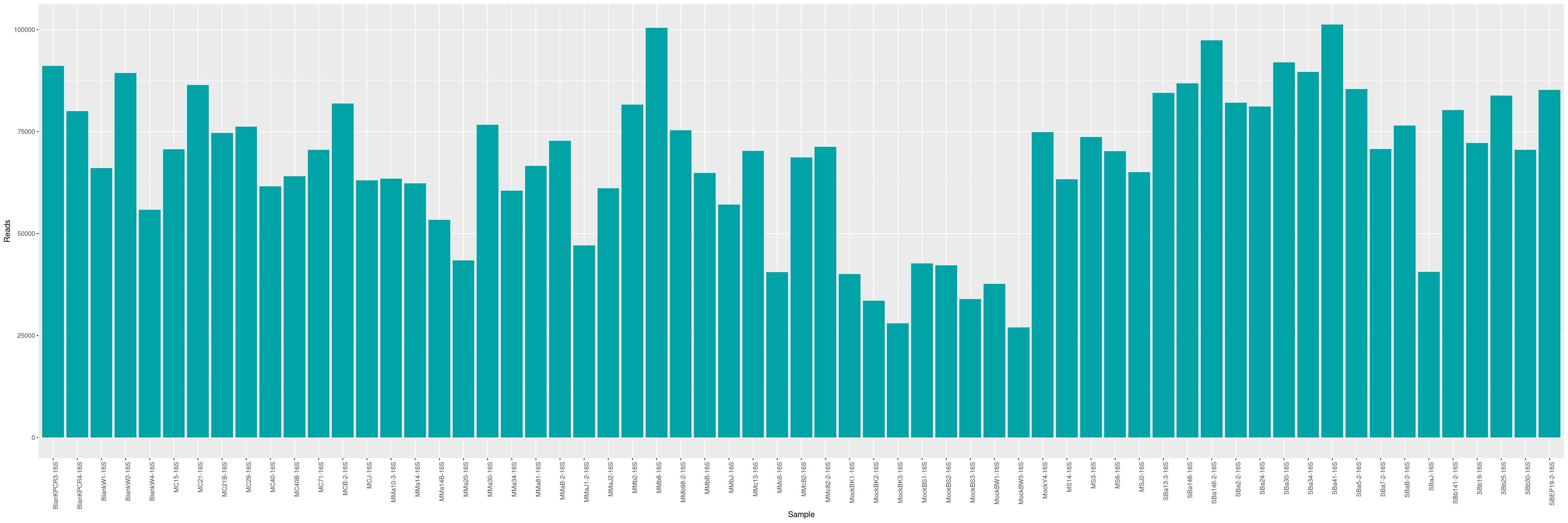
cd /home/orue/work/PROJECTS/LTMCBW/ITS/
mkdir LOGS
qsub -cwd -V -e LOGS -o LOGS -N seqkit -pe thread 4 -R y -b y "conda activate seqkit-2.0.0 && seqkit stats /home/orue/work/PROJECTS/LTMCBW/ITS/*.fastq.gz -j 4 > raw_data.infos && conda deactivate"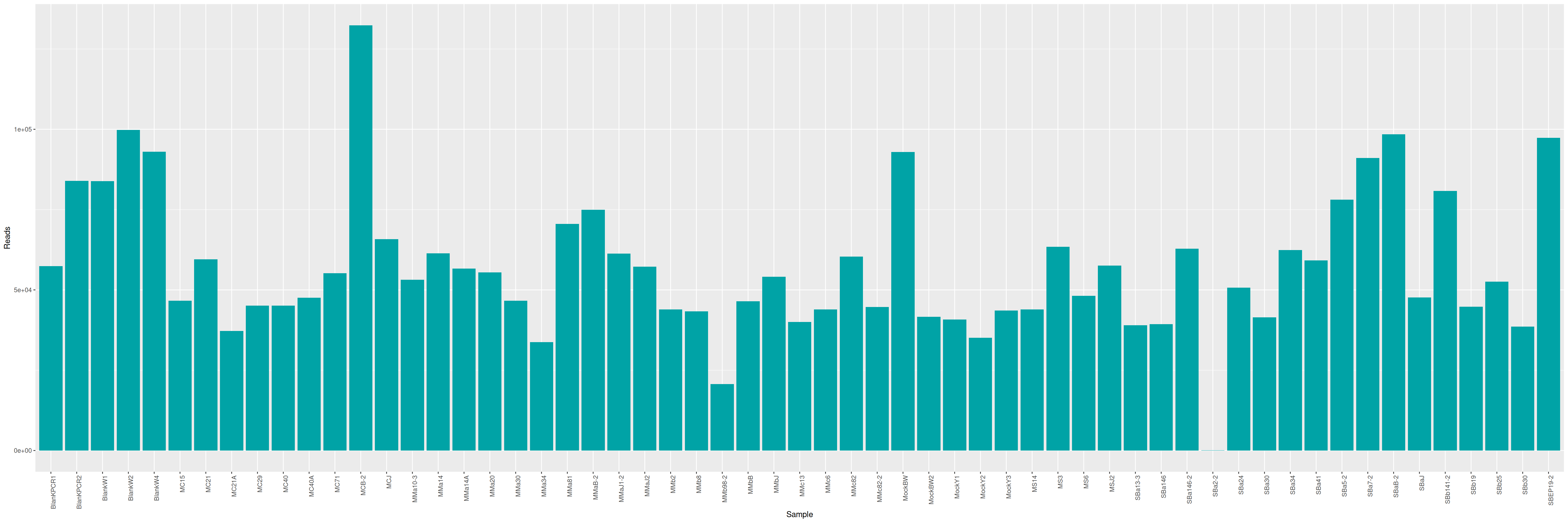
Warning
Some unexpected metrics are observed: the length of the reads is not homogeneous and the average length is relatively low for a classic Illumina run.
cd /home/orue/work/PROJECTS/LTMCBW/16S
mkdir FASTQC
for i in /home/orue/work/PROJECTS/LTMCBW/16S/*.fastq.gz ; do echo "conda activate fastqc-0.11.9 && fastqc $i -o FASTQC && conda deactivate" >> fastqc.sh ; done
qarray -cwd -V -N fastqc -o LOGS -e LOGS fastqc.sh
qsub -cwd -V -N multiqc -o LOGS -e LOGS -b y "conda activate multiqc-1.11 && multiqc FASTQC -o MULTIQC && conda deactivate"The MultiQC report shows that base qualities are moderately good, and a large proportion of reads are small.
cd /home/orue/work/PROJECTS/LTMCBW/ITS
mkdir FASTQC
for i in /home/orue/work/PROJECTS/LTMCBW/ITS/*.fastq.gz ; do echo "conda activate fastqc-0.11.9 && fastqc $i -o FASTQC && conda deactivate" >> fastqc.sh ; done
qarray -cwd -V -N fastqc -o LOGS -e LOGS fastqc.sh
qsub -cwd -V -N multiqc -o LOGS -e LOGS -b y "conda activate multiqc-1.11 && multiqc FASTQC -o MULTIQC && conda deactivate"The MultiQC report shows that base qualities are moderately good, and a large proportion of reads are small.
Warning
Although the quality of the reads is relatively poor, this is due to the difficulty of extracting the DNA. Consequently, a lot of reads are small. They are surely not contiguous with such small sizes.
Bioinformatics
We used FROGS
The first tool, called preprocess allows to clean reads. From FASTQ files, reads with N were first discarded. Then, reads were denoised with dada2
cd /home/orue/work/PROJECTS/LTMCBW/16S/
tar zcvf 16S.tar.gz *.fastq.gz
mkdir FROGS5
cd FROGS5
conda activate frogs-4.1.0
conda_lib_dir=`echo $(dirname $(dirname $(which preprocess.py)))/libexec`
export PATH=$conda_lib_dir:$PATH
export PATH="/home/orue/work/GIT/FROGS2023OK/libexec":$PATH
export PATH="/home/orue/work/GIT/FROGS2023OK/app":$PATH
export PYTHONPATH=`echo $(dirname $(dirname $(which preprocess.py)))/lib`:$PYTHONPATH
denoising.py illumina --min-amplicon-size 300 --max-amplicon-size 590 --merge-software pear --five-prim-primer TACGGRAGGCAGCAG --three-prim-primer AGGATTAGATACCCTGGTA --R1-size 300 --R2-size 300 --nb-cpus 16 --output-fasta clusters.fasta --output-biom clusters.biom --summary denoising.html --log-file denoising.log --process dada2 --input-archive ../16S.tar.gz
Warning
As expected, a lot of 16S reads are lost because they are not contiguous.
cd /home/orue/work/PROJECTS/LTMCBW/ITS/
tar zcvf ITS.tar.gz *.fastq.gz
mkdir FROGS5
cd FROGS5
conda activate frogs-4.1.0
conda_lib_dir=`echo $(dirname $(dirname $(which preprocess.py)))/libexec`
export PATH=$conda_lib_dir:$PATH
export PATH="/home/orue/work/GIT/FROGS2023OK/libexec":$PATH
export PATH="/home/orue/work/GIT/FROGS2023OK/app":$PATH
export PYTHONPATH=`echo $(dirname $(dirname $(which preprocess.py)))/lib`:$PYTHONPATH
denoising.py illumina --min-amplicon-size 50 --max-amplicon-size 1000 --merge-software pear --five-prim-primer GCATCGATGAAGAACGCAGC --three-prim-primer GCAWAWCAAWAAGCGGAGGA --R1-size 300 --R2-size 300 --nb-cpus 16 --output-fasta clusters.fasta --output-biom clusters.biom --summary preprocess.html --log-file preprocess.log --process dada2 --input-archive ../ITS.tar.gz --keep-unmergedThe affiliation with a databank constituted of UNITE v.9.0 and METABARFOOD sequences was performed from the ITSx BIOM and FASTA files. The databank build is described here.
conda activate frogs-4.1.0
cd /home/orue/work/PROJECTS/LTMCBW/ITS
cp /projet/gxyprod/galaxy/database/files/000/759/dataset_759593.dat itsx.biom
cp /projet/gxyprod/galaxy/database/files/000/759/dataset_759592.dat itsx.fasta
ln -s ~/work/PROJECTS/LEBANESEWHEATSOURDOUGH/ITS/UNITE_9.0_20221016_plus_METABARFOOD.fasta* .
taxonomic_affiliation.py --reference UNITE_9.0_20221016_plus_METABARFOOD.fasta --input-biom itsx.biom --input-fasta itsx.fasta --output-biom affiliation.biom --nb-cpus 8 --taxonomy-ranks 'Domain', 'Phylum', 'Class', 'Order', 'Family', 'Genus', 'Species'
biom_to_tsv.py --input-biom affiliation.biom --output-tsv affiliation.tsvJust to check the taxonomies:
biomfile <- "html/affiliation.biom"
frogs <- import_frogs(biomfile, taxMethod = "blast")
data <- data.frame("Name" = rep(1:length(sample_names(frogs))))
rownames(data) <- sample_names(frogs)
sample_data(frogs) <- data
plot_composition(frogs, taxaRank1 = NULL, taxaSet1 = NULL, taxaRank2 = "Genus", numberOfTaxa = 20)Problematic taxa
taxa Kingdom Phylum Class
Cluster_8954 Cluster_8954 Fungi Ascomycota Saccharomycetes
Cluster_8953 Cluster_8953 Fungi Ascomycota Saccharomycetes
Cluster_9329 Cluster_9329 Fungi Multi-affiliation Multi-affiliation
Order Family Genus rank
Cluster_8954 Saccharomycetales Pichiaceae Pichia 6
Cluster_8953 Saccharomycetales Saccharomycetaceae Pichia 8
Cluster_9329 Multi-affiliation Multi-affiliation Multi-affiliation 11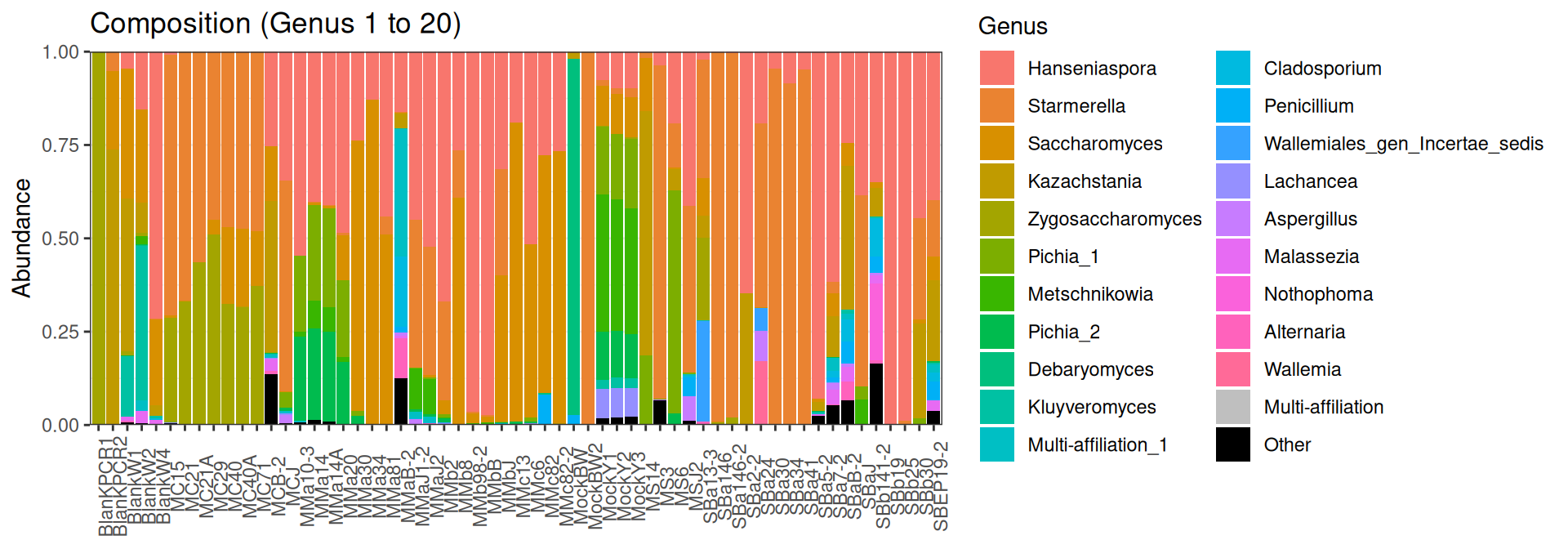
Deliverables agreed at the preliminary meeting (Table 2).
| Definition | Result | |
|---|---|---|
| 1 | HTML report | https://documents.migale.inrae.fr/posts/analyses/ltmcbw-84512/ |
| 2 | Archive containing data to be stored | not yet |
| 3 | BIOM and FASTA out from FROGS5 preprocess | Galaxy histories : LTMCBW ITS FROGS5 and LTMCBW 16S FROGS5 |
| 4 | BIOM file after affiliation with METABARFOOD and UNITE sequences | Deposited in Galaxy history |
LTMCBW2
Raw data
cd /home/orue/work/PROJECTS/LTMCBW2/
mkdir RAW_DATA_READY
# Tri des échantillons entre ceux de Paul et Pamela
# Renommage des fichiers
rename -n 's/_[.*]_//; s/_001//' *.fastq.gz
rename 's/_.*_/_/' *.fastq.gz
mkdir 16S
mkdir ITS
mv *-16S* 16S/
mv *-ITS* ITS/
mv Bla* 16S/
cd 16S/
tar zcvf LTMCBW2_16S.tar.gz *.fastq.gz
cd ../ITS/
tar zcvf LTMCBW2_ITS.tar.gz *.fastq.gzQuality control
cd /home/orue/work/PROJECTS/LTMCBW2/RAW_DATA_READY/16S/
mkdir LOGS
qsub -cwd -V -e LOGS -o LOGS -N seqkit -pe thread 4 -R y -b y "conda activate seqkit-2.0.0 && seqkit stats /home/orue/work/PROJECTS/LTMCBW2/RAW_DATA_READY/16S/*.fastq.gz -j 4 > raw_data.infos && conda deactivate"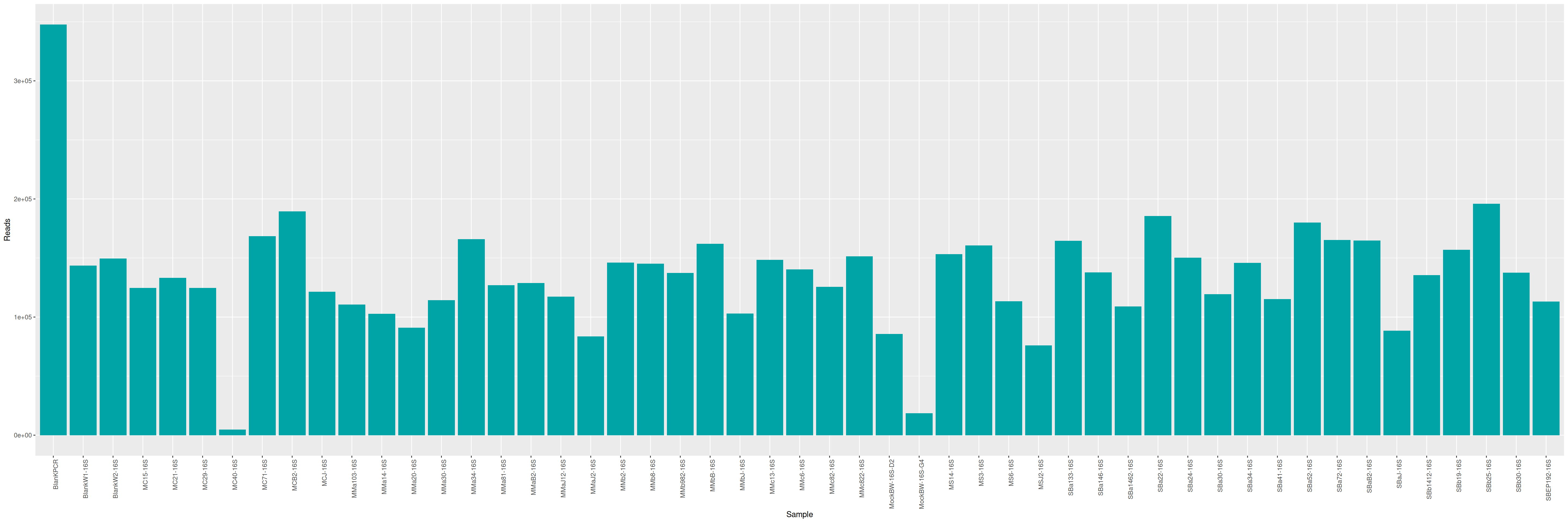
cd /home/orue/work/PROJECTS/LTMCBW2/RAW_DATA_READY/16S
mkdir FASTQC
for i in /home/orue/work/PROJECTS/LTMCBW2/RAW_DATA_READY/16S/*.fastq.gz ; do echo "conda activate fastqc-0.11.9 && fastqc $i -o FASTQC && conda deactivate" >> fastqc.sh ; done
qarray -cwd -V -N fastqc -o LOGS -e LOGS fastqc.sh
qsub -cwd -V -N multiqc -o LOGS -e LOGS -b y "conda activate multiqc-1.11 && multiqc FASTQC -o MULTIQC && conda deactivate"The MultiQC report shows that base qualities are moderately good, and a large proportion of reads are small.
#cd /home/orue/work/PROJECTS/LTMCBW/ITS
#mkdir FASTQC
#for i in /home/orue/work/PROJECTS/LTMCBW/ITS/*.fastq.gz ; do echo "conda activate fastqc-0.11.9 && fastqc $i -o FASTQC && conda deactivate" >> fastqc.sh ; done
#qarray -cwd -V -N fastqc -o LOGS -e LOGS fastqc.sh
#qsub -cwd -V -N multiqc -o LOGS -e LOGS -b y "conda activate multiqc-1.11 && multiqc FASTQC -o MULTIQC && conda deactivate"The MultiQC report shows that base qualities are moderately good, and a large proportion of reads are small.
Bioinformatics
cd /home/orue/work/PROJECTS/LTMCBW2/RAW_DATA_READY/16S
mkdir FROGS5-W-PRIMER
cd FROGS5-W-PRIMER
#conda activate frogs-4.1.0
#conda_lib_dir=`echo $(dirname $(dirname $(which denoising.py)))/libexec`
#export PATH=$conda_lib_dir:$PATH
#export PATH="/home/orue/work/GIT/FROGS2023OK/libexec":$PATH
#export PATH="/home/orue/work/GIT/FROGS2023OK/app":$PATH
#export PYTHONPATH=`echo $(dirname $(dirname $(which denoising.py)))/lib`:$PYTHONPATH
conda activate frogs-5.0.0
denoising.py illumina --min-amplicon-size 300 --max-amplicon-size 590 --merge-software pear --five-prim-primer TACGGRAGGCWGCAG --three-prim-primer GGATTAGATACCCBDGTAGTC --R1-size 300 --R2-size 300 --nb-cpus 16 --output-fasta clusters.fasta --output-biom clusters.biom --html denoising.html --log-file denoising.log --process dada2 --input-archive ../LTMCBW2_16S.tar.gz
conda deactivatecd /home/orue/work/PROJECTS/LTMCBW2/RAW_DATA_READY/ITS/
mkdir FROGS5
cd FROGS5
conda activate frogs-4.1.0
conda_lib_dir=`echo $(dirname $(dirname $(which denoising.py)))/libexec`
export PATH=$conda_lib_dir:$PATH
export PATH="/home/orue/work/GIT/FROGS2023OK/libexec":$PATH
export PATH="/home/orue/work/GIT/FROGS2023OK/app":$PATH
export PYTHONPATH=`echo $(dirname $(dirname $(which denoising.py)))/lib`:$PYTHONPATH
denoising.py illumina --min-amplicon-size 50 --max-amplicon-size 1000 --merge-software pear --five-prim-primer GCATCGATGAAGAACGCAGC --three-prim-primer GCAWAWCAAWAAGCGGAGGA --R1-size 300 --R2-size 300 --nb-cpus 16 --output-fasta clusters.fasta --output-biom clusters.biom --html denoising.html --log-file preprocess.log --process dada2 --input-archive ../LTMCBW2_ITS.tar.gz --keep-unmerged
remove_chimera.py --input-biom clusters.biom --input-fasta clusters.fasta --html remove_chimera.html --log-file remove_chimera.log
cluster_filters.py --input-biom remove_chimera_abundance.biom --input-fasta remove_chimera.fasta --min-abundance 5e-05 --nb-cpus 16 --html cluster_filters.html
itsx.py --input-biom cluster_filters_abundance.biom --input-fasta cluster_filters.fasta --nb-cpus 8 --organism-groups F --check-its-only --html itsx.html --log-file itsx.log
ln -s /home/orue/work/PROJECTS/LEBANESEWHEATSOURDOUGH/ITS/UNITE_9.0_20221016_plus_METABARFOOD.fasta* .
taxonomic_affiliation.py --reference UNITE_9.0_20221016_plus_METABARFOOD.fasta --input-biom itsx_abundance.biom --input-fasta itsx.fasta --output-biom affiliation.biom --nb-cpus 8 --taxonomy-ranks 'Domain', 'Phylum', 'Class', 'Order', 'Family', 'Genus', 'Species'
biom_to_tsv.py --input-biom affiliation.biom --output-tsv affiliation.tsvComparison of ITS between both runs
common_samples <- c("MCB-2","MMb98-2","MS3","SBa13-3","SBa146-2","SBa2-2","SBa34","SBa7-2","SBaB-2")
# 1er run
biomfile1 <- "html/affiliation.biom"
frogs1 <- import_frogs(biomfile1, taxMethod = "blast")
data <- data.frame("Name" = str_replace(sample_names(frogs1),"-ITS",""), "Run" = 1)
rownames(data) <- sample_names(frogs1)
sample_data(frogs1) <- data
#frogs1 <- prune_samples(samples = common_samples, x = frogs1)
#####sample_data(frogs1)$Name <- sample_names()
#frogs1
sample_names(frogs1) <- glue::glue("{sample_names(frogs1)}_run1")
#taxa_names(frogs1) <- paste(taxa_names(frogs1), "1", sep = "_")
frogs1phyloseq-class experiment-level object
otu_table() OTU Table: [ 278 taxa and 59 samples ]
sample_data() Sample Data: [ 59 samples by 2 sample variables ]
tax_table() Taxonomy Table: [ 278 taxa by 7 taxonomic ranks ]# 2nd run
biomfile2 <- "data2/affiliation_ITS.biom"
frogs2 <- import_frogs(biomfile2, taxMethod = "blast")
data <- data.frame("Name" = str_replace(sample_names(frogs2),"-ITS",""), "Run" = 2)
data$Name <- common_samples
rownames(data) <- sample_names(frogs2)
sample_data(frogs2) <- data
sample_names(frogs2) <- common_samples
frogs2phyloseq-class experiment-level object
otu_table() OTU Table: [ 315 taxa and 9 samples ]
sample_data() Sample Data: [ 9 samples by 2 sample variables ]
tax_table() Taxonomy Table: [ 315 taxa by 7 taxonomic ranks ]sample_names(frogs2) <- glue::glue("{sample_names(frogs2)}_run2")
taxa_names(frogs2) <- paste(taxa_names(frogs2), "2", sep = "_")
frogs2phyloseq-class experiment-level object
otu_table() OTU Table: [ 315 taxa and 9 samples ]
sample_data() Sample Data: [ 9 samples by 2 sample variables ]
tax_table() Taxonomy Table: [ 315 taxa by 7 taxonomic ranks ]frogs_merge <- merge_phyloseq(frogs1, frogs2)
saveRDS(frogs_merge,"html2/bothRuns.rds")
sub <- subset_samples(frogs_merge, Name %in% common_samples)
saveRDS(sub, "html2/9samples.rds")The compositions between samples sequenced twice are not very similar…
p <- plot_composition(sub, taxaRank1 = NULL, taxaSet1 = NULL, taxaRank2 = "Family", numberOfTaxa = 20)Problematic taxa
taxa Kingdom Phylum Class
Cluster_11_2 Cluster_11_2 Fungi Multi-affiliation Multi-affiliation
Order Family rank
Cluster_11_2 Multi-affiliation Multi-affiliation 9p + facet_grid(~Name, scales = "free_x")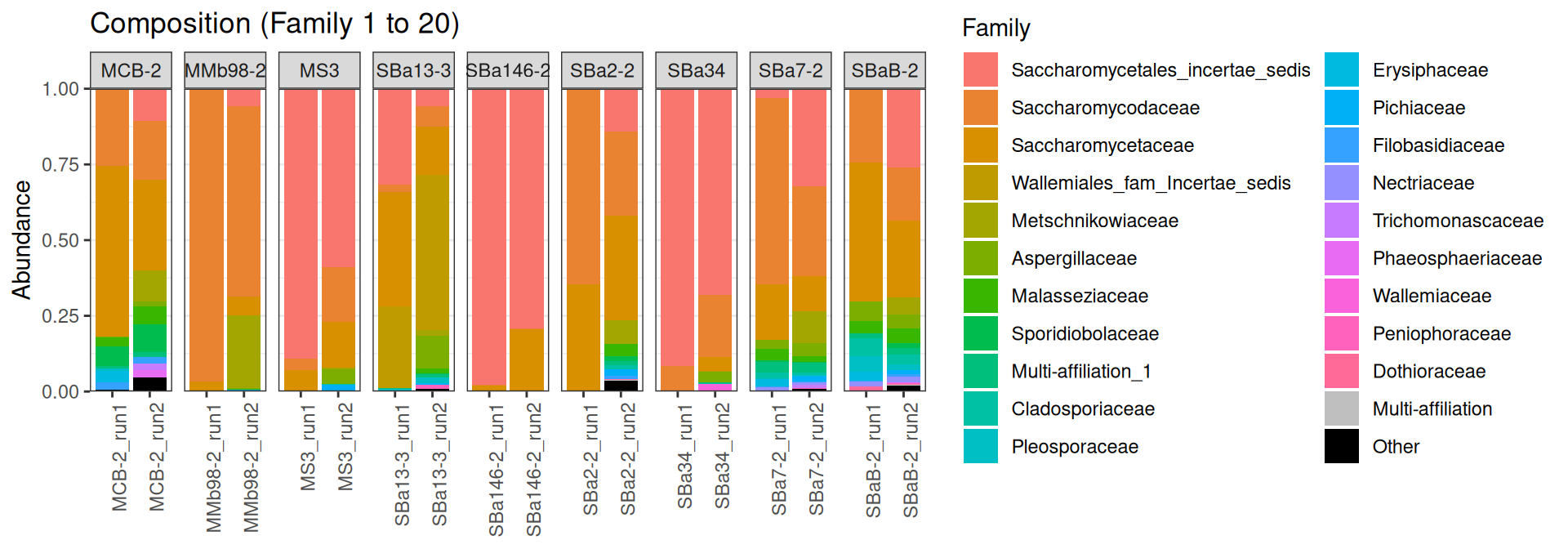
Merge of both runs
I think it would be better to replace all 8 samples with run 2 but keep SBa146-2 of run1 if it’s possible ?
frogs_mergephyloseq-class experiment-level object
otu_table() OTU Table: [ 593 taxa and 68 samples ]
sample_data() Sample Data: [ 68 samples by 2 sample variables ]
tax_table() Taxonomy Table: [ 593 taxa by 7 taxonomic ranks ]sample_names(frogs_merge) [1] "BlanKPCR1_run1" "BlanKPCR2_run1" "BlankW1_run1" "BlankW2_run1"
[5] "BlankW4_run1" "MC15_run1" "MC21A_run1" "MC21_run1"
[9] "MC29_run1" "MC40A_run1" "MC40_run1" "MC71_run1"
[13] "MCB-2_run1" "MCJ_run1" "MMa10-3_run1" "MMa14A_run1"
[17] "MMa14_run1" "MMa20_run1" "MMa30_run1" "MMa34_run1"
[21] "MMa81_run1" "MMaB-2_run1" "MMaJ1-2_run1" "MMaJ2_run1"
[25] "MMb2_run1" "MMb8_run1" "MMb98-2_run1" "MMbB_run1"
[29] "MMbJ_run1" "MMc13_run1" "MMc6_run1" "MMc82-2_run1"
[33] "MMc82_run1" "MS14_run1" "MS3_run1" "MS6_run1"
[37] "MSJ2_run1" "MockBW2_run1" "MockBW_run1" "MockY1_run1"
[41] "MockY2_run1" "MockY3_run1" "SBEP19-2_run1" "SBa13-3_run1"
[45] "SBa146-2_run1" "SBa146_run1" "SBa2-2_run1" "SBa24_run1"
[49] "SBa30_run1" "SBa34_run1" "SBa41_run1" "SBa5-2_run1"
[53] "SBa7-2_run1" "SBaB-2_run1" "SBaJ_run1" "SBb141-2_run1"
[57] "SBb19_run1" "SBb25_run1" "SBb30_run1" "MCB-2_run2"
[61] "MMb98-2_run2" "MS3_run2" "SBa13-3_run2" "SBa146-2_run2"
[65] "SBa2-2_run2" "SBa34_run2" "SBa7-2_run2" "SBaB-2_run2" samples_to_remove <- c("MCB-2_run1", "MMb98-2_run1", "MS3_run1", "SBa13-3_run1", "SBa146-2_run2", "SBa2-2_run1", "SBa34_run1","SBa7-2_run1", "SBaB-2_run1")
frogs_merge_final <- prune_samples(!(sample_names(frogs_merge) %in% samples_to_remove), frogs_merge)
frogs_merge_finalphyloseq-class experiment-level object
otu_table() OTU Table: [ 593 taxa and 59 samples ]
sample_data() Sample Data: [ 59 samples by 2 sample variables ]
tax_table() Taxonomy Table: [ 593 taxa by 7 taxonomic ranks ]sample_data(frogs_merge) Name Run
BlanKPCR1_run1 BlanKPCR1 1
BlanKPCR2_run1 BlanKPCR2 1
BlankW1_run1 BlankW1 1
BlankW2_run1 BlankW2 1
BlankW4_run1 BlankW4 1
MC15_run1 MC15 1
MC21A_run1 MC21A 1
MC21_run1 MC21 1
MC29_run1 MC29 1
MC40A_run1 MC40A 1
MC40_run1 MC40 1
MC71_run1 MC71 1
MCB-2_run1 MCB-2 1
MCJ_run1 MCJ 1
MMa10-3_run1 MMa10-3 1
MMa14A_run1 MMa14A 1
MMa14_run1 MMa14 1
MMa20_run1 MMa20 1
MMa30_run1 MMa30 1
MMa34_run1 MMa34 1
MMa81_run1 MMa81 1
MMaB-2_run1 MMaB-2 1
MMaJ1-2_run1 MMaJ1-2 1
MMaJ2_run1 MMaJ2 1
MMb2_run1 MMb2 1
MMb8_run1 MMb8 1
MMb98-2_run1 MMb98-2 1
MMbB_run1 MMbB 1
MMbJ_run1 MMbJ 1
MMc13_run1 MMc13 1
MMc6_run1 MMc6 1
MMc82-2_run1 MMc82-2 1
MMc82_run1 MMc82 1
MS14_run1 MS14 1
MS3_run1 MS3 1
MS6_run1 MS6 1
MSJ2_run1 MSJ2 1
MockBW2_run1 MockBW2 1
MockBW_run1 MockBW 1
MockY1_run1 MockY1 1
MockY2_run1 MockY2 1
MockY3_run1 MockY3 1
SBEP19-2_run1 SBEP19-2 1
SBa13-3_run1 SBa13-3 1
SBa146-2_run1 SBa146-2 1
SBa146_run1 SBa146 1
SBa2-2_run1 SBa2-2 1
SBa24_run1 SBa24 1
SBa30_run1 SBa30 1
SBa34_run1 SBa34 1
SBa41_run1 SBa41 1
SBa5-2_run1 SBa5-2 1
SBa7-2_run1 SBa7-2 1
SBaB-2_run1 SBaB-2 1
SBaJ_run1 SBaJ 1
SBb141-2_run1 SBb141-2 1
SBb19_run1 SBb19 1
SBb25_run1 SBb25 1
SBb30_run1 SBb30 1
MCB-2_run2 MCB-2 2
MMb98-2_run2 MMb98-2 2
MS3_run2 MS3 2
SBa13-3_run2 SBa13-3 2
SBa146-2_run2 SBa146-2 2
SBa2-2_run2 SBa2-2 2
SBa34_run2 SBa34 2
SBa7-2_run2 SBa7-2 2
SBaB-2_run2 SBaB-2 2saveRDS(frogs_merge_final, "html2/final_physeq_59samples.rds")Archive with the 59 samples and run denoising
cd cd ~/work/PROJECTS/LTMCBW2/RAW_DATA_READY/ITS/RUN1_AND_2/
mkdir 59samples
cd 59samples/
cp ../*.fastq.gz .
rm -f MCB-2_R*
rm -f MMb98-2_R*
rm -f MS3_R*
rm -f SBa13-3_R*
rm -f SBa146-2_R*
rm -f SBa2-2_R*
rm -f SBa34_R*
rm -f SBa7-2_R*
rm -f SBaB-2_R*
for i in *_R1.fastq.gz ; do echo $i ; done |wc -l
59
tar zcvf 59samplesITS.tar.gz *.fastq.gz
conda activate frogs-4.1.0
conda_lib_dir=`echo $(dirname $(dirname $(which denoising.py)))/libexec`
export PATH=$conda_lib_dir:$PATH
export PATH="/home/orue/work/GIT/FROGS2023OK/libexec":$PATH
export PATH="/home/orue/work/GIT/FROGS2023OK/app":$PATH
export PYTHONPATH=`echo $(dirname $(dirname $(which denoising.py)))/lib`:$PYTHONPATH
denoising.py illumina --min-amplicon-size 300 --max-amplicon-size 590 --merge-software pear --five-prim-primer TACGGRAGGCAGCAG --three-prim-primer GGATTAGATACCCBDGTAGTC --R1-size 300 --R2-size 300 --nb-cpus 16 --output-fasta clusters.fasta --output-biom clusters.biom --html denoising.html --log-file denoising.log --process dada2 --input-archive 59samplesITS.tar.gz Then, the affiliation was performed with the METABARFOOD_UNITE reference databank after getting files from Pamela Galaxy history:
taxonomic_affiliation.py --reference ../UNITE_9.0_20221016_plus_METABARFOOD.fasta --input-biom taxonomic_affiliation_pamela.biom --input-fasta taxonomic_affiliation_pamela.fasta --output-biom affiliation.biom --nb-cpus 8 --taxonomy-ranks 'Domain', 'Phylum', 'Class', 'Order', 'Family', 'Genus', 'Species'Downloads
References
1. Shen W, Le S, Li Y, Hu F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/q file manipulation. PloS one. 2016;11:e0163962.
2. Andrews S. FastQC a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
3. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–8.
4. Escudié F, Auer L, Bernard M, Mariadassou M, Cauquil L, Vidal K, et al. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics. 2018;34:1287–94. doi:10.1093/bioinformatics/btx791.
5. Bernard M, Rué O, Mariadassou M, Pascal G. FROGS: a powerful tool to analyse the diversity of fungi with special management of internal transcribed spacers. Briefings in Bioinformatics. 2021;22. doi:10.1093/bib/bbab318.
6. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from illumina amplicon data. Nature methods. 2016;13:581.
7. Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: A fast and accurate illumina paired-end reAd mergeR. Bioinformatics. 2013;30:614–20.
8. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet journal. 2011;17:10–2.
Reuse
This document will not be accessible without prior agreement of the partners
