
mkdir /home/orue/work/PROJECTS/HOLOVINI/
cd /home/orue/work/PROJECTS/HOLOVINI/
mkdir -p 16S/RAW_DATA/ ITS/RAW_DATA/
cd 16S
cp ../../LTMCBW2/RAW_DATA_READY/PAUL/*16S* .
rename 's/-16S_S\d+_/_/' *.fastq.gz
mkdir RAW_DATA/
mv *.fastq.gz RAW_DATA/
cp ../../LTMCBW2/RAW_DATA_READY/16S/MockBW-16S-*
mv MockBW-16S-D2_R2.fastq.gz RAW_DATA/MockBW-D2_R2.fastq.gz
mv MockBW-16S-D2_R1.fastq.gz RAW_DATA/MockBW-D2_R1.fastq.gz
mv MockBW-16S-G4_R2.fastq.gz RAW_DATA/MockBW-G4_R2.fastq.gz
mv MockBW-16S-G4_R1.fastq.gz RAW_DATA/MockBW-G4_R1.fastq.gz
for i in RAW_DATA/*_R1.fastq.gz ; do echo $i ; done |wc -l
35 # 16S samples
cd ../ITS
cp ../../LTMCBW2/RAW_DATA_READY/PAUL/*ITS* .
rename 's/-ITS_S\d+_/_/' *.fastq.gz
mv *.fastq.gz RAW_DATA/
cp ../../LTMCBW2/RAW_DATA_READY/PAUL/MockY-Cave_S92_R* .
renamed 'MockY-Cave_S92_R2.fastq.gz' -> 'RAW_DATA/MockY-Cave_R2.fastq.gz'
mv MockY-Cave_S92_R1.fastq.gz RAW_DATA/MockY-Cave_R1.fastq.gz
mv MockY-Cave_S92_R2.fastq.gz RAW_DATA/MockY-Cave_R2.fastq.gz
for i in RAW_DATA/*_R1.fastq.gz ; do echo $i ; done |wc -l
35 # ITS samples
cd ../
scp ITS/RAW_DATA/*.fastq.gz orue@abaca.maiage.inrae.fr:/backup/partage/migale/HOLOVINI/ITS/
scp 16S/RAW_DATA/*.fastq.gz orue@abaca.maiage.inrae.fr:/backup/partage/migale/HOLOVINI/16S/
Note
This document is a report of the analyses performed. You will find all the code used to analyze these data. The version of the tools (maybe in code chunks) and their references are indicated, for questions of reproducibility.
Aim of the project
As part of the HOLOVINI project funded by the HOLOFLUX metaprogramme, we aim to identify microbial flows between the environment, insects, vines and winery.
Partners
- Olivier Rué - Migale bioinformatics facility - BioInfomics - INRAE
- Jean-Luc Legras - SPO - INRAE
Deliverables
Deliverables agreed at the preliminary meeting (Table 1).
| Definition | Status | |
|---|---|---|
| 1 | HTML report | ✔️ |
| 2 | RDS files | ✔️ |
| 3 | Taxonomies and abundances for a manual curation | ✔️ |
Data management
Important
All data is managed by the migale facility for the duration of the project. Once the project is over, the Migale facility does not keep your data. We will provide you with the raw data and associated metadata that will be deposited on public repositories before the results are used. We can guide you in the submission process. We will then decide which files to keep, knowing that this report will also be provided to you and that the analyses can be replayed if needed.
Raw data
Raw data were available with those of the project LTMCBW2, available on the front server. A copy was sent to the abaca server.
Quality control
# seqkit
cd /home/orue/work/PROJECTS/HOLOVINI/16S
qsub -cwd -V -N seqkit -pe thread 4 -R y -b y "conda activate seqkit-2.0.0 && seqkit stats /home/orue/work/PROJECTS/HOLOVINI/16S/RAW_DATA/*.fastq.gz -j 4 > raw_data.infos && conda deactivate"We can plot and display the number of reads to see if enough reads are present and if samples are homogeneous.
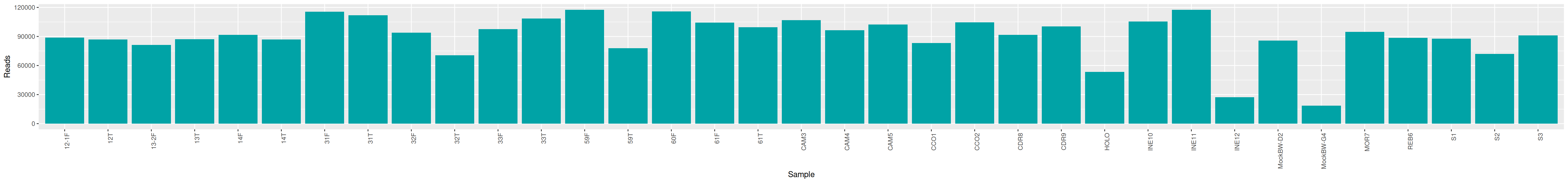
raw_data %>% select(sample, num_seqs, sum_len, min_len, avg_len, max_len) %>% datatable()cd /home/orue/work/PROJECTS/HOLOVINI/16S/
mkdir FASTQC LOGS
for i in /home/orue/work/PROJECTS/HOLOVINI/16S/RAW_DATA/*.fastq.gz ; do echo "conda activate fastqc-0.11.9 && fastqc $i -o FASTQC && conda deactivate" >> fastqc.sh ; done
qarray -cwd -V -N fastqc -o LOGS -e LOGS fastqc.sh
qsub -cwd -V -N multiqc -o LOGS -e LOGS -b y "conda activate multiqc-1.11 && multiqc FASTQC -o MULTIQC && conda deactivate"The MultiQC report shows expected metrics for Illumina Miseq sequencing data.
Note
The quality control is good enough to go further.
# seqkit
cd /home/orue/work/PROJECTS/HOLOVINI/ITS
qsub -cwd -V -N seqkit -pe thread 4 -R y -b y "conda activate seqkit-2.0.0 && seqkit stats /home/orue/work/PROJECTS/HOLOVINI/ITS/RAW_DATA/*.fastq.gz -j 4 > raw_data.infos && conda deactivate"We can plot and display the number of reads to see if enough reads are present and if samples are homogeneous.
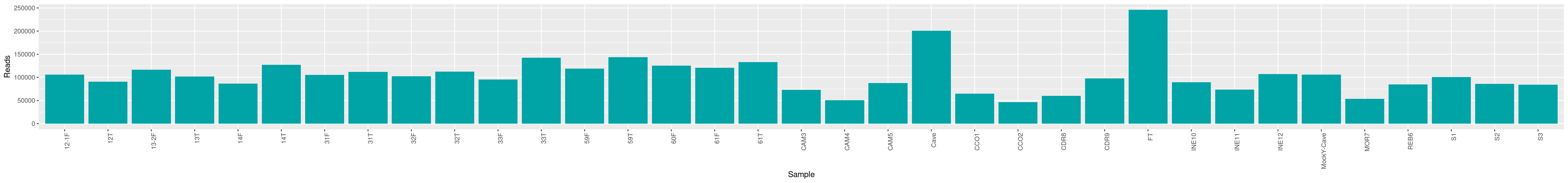
raw_data %>% select(sample, num_seqs, sum_len, min_len, avg_len, max_len) %>% datatable()cd /home/orue/work/PROJECTS/HOLOVINI/ITS/
mkdir FASTQC LOGS
for i in /home/orue/work/PROJECTS/HOLOVINI/ITS/RAW_DATA/*.fastq.gz ; do echo "conda activate fastqc-0.11.9 && fastqc $i -o FASTQC && conda deactivate" >> fastqc.sh ; done
qarray -cwd -V -N fastqc -o LOGS -e LOGS fastqc.sh
qsub -cwd -V -N multiqc -o LOGS -e LOGS -b y "conda activate multiqc-1.11 && multiqc FASTQC -o MULTIQC && conda deactivate"The MultiQC report shows expected metrics for Illumina Miseq sequencing data.
Note
The quality control is good enough to go further.
Bioinformatics
We used FROGS v.5.0.0
The first tool, called denoising allows to clean reads. From FASTQ files, reads with N were first discarded. Then, reads were denoised with dada2
cd /home/orue/work/PROJECTS/HOLOVINI/16S
cd RAW_DATA/
tar zcvf Holovini_16S.tar.gz *.fastq.gz
cd ../
mkdir FROGS5
cd FROGS5
conda activate frogs-5.0.0
denoising.py illumina --min-amplicon-size 300 --max-amplicon-size 590 --merge-software pear --five-prim-primer TACGGRAGGCAGCAG --three-prim-primer GGATTAGATACCCBDGTAGTC --R1-size 300 --R2-size 300 --nb-cpus 16 --output-fasta clusters.fasta --output-biom clusters.biom --html denoising.html --log-file denoising.log --process dada2 --input-archive ../RAW_DATA/Holovini_16S.tar.gzremove_chimera.py --input-biom clusters.biom --input-fasta clusters.fasta --html remove_chimera.html --log-file remove_chimera.log --nb-cpus 1620% of sequences detected as chimera and removed
cluster_filters.py --input-fasta remove_chimera.fasta --input-biom remove_chimera_abundance.biom --nb-cpus 16 --contaminant /db/outils/FROGS/contaminants/phi.fa --min-abundance 0.00005 --output-fasta filters.fasta --log-file filters.log --html cluster_filters.htmltaxonomic_affiliation.py --input-biom cluster_filters_abundance.biom --input-fasta filters.fasta --nb-cpus 16 --reference /db/outils/FROGS/assignation/silva_138.1_16S_pintail100/silva_138.1_16S_pintail100.fasta --log-file taxonomic_affiliation.logtree.py --input-biom affiliation_abundance.biom --html tree.html --output-tree tree.nwk --log-file tree.log --input-fasta filters.fastacd /home/orue/work/PROJECTS/HOLOVINI/ITS
cd RAW_DATA/
tar zcvf Holovini_ITS.tar.gz *.fastq.gz
cd ../
mkdir FROGS5
cd FROGS5
conda activate frogs-5.0.0
denoising.py illumina --min-amplicon-size 50 --max-amplicon-size 1000 --merge-software pear --five-prim-primer GCATCGATGAAGAACGCAGC --three-prim-primer GCAWAWCAAWAAGCGGAGGA --R1-size 300 --R2-size 300 --nb-cpus 16 --output-fasta clusters.fasta --output-biom clusters.biom --html denoising.html --log-file preprocess.log --process dada2 --input-archive ../RAW_DATA/Holovini_ITS.tar.gz --keep-unmergedremove_chimera.py --input-biom clusters.biom --input-fasta clusters.fasta --html remove_chimera.html --log-file remove_chimera.log --nb-cpus 16cluster_filters.py --input-fasta remove_chimera.fasta --input-biom remove_chimera_abundance.biom --nb-cpus 16 --contaminant /db/outils/FROGS/contaminants/phi.fa --min-abundance 0.00005 --output-fasta filters.fasta --log-file filters.log --html cluster_filters.htmlitsx.py --check-its-only --input-fasta filters.fasta --input-biom cluster_filters_abundance.biom --log-file itsx.logtaxonomic_affiliation.py --input-biom itsx_abundance.biom --input-fasta itsx.fasta --nb-cpus 16 --reference ~/work/PROJECTS/LEBANESEWHEATSOURDOUGH/ITS/UNITE_9.0_20221016_plus_METABARFOOD.fasta --log-file taxonomic_affiliation.logtree.py --input-biom affiliation_abundance.biom --html tree.html --output-tree tree.nwk --log-file tree.log --input-fasta itsx.fastaBiostatisctics
Phyloseq [9] is a R package dedicated to diversity analyses.
To create a phyloseq object, we need the BIOM file, the metadata file and eventually a tree file.
library(phyloseq)
library(phyloseq.extended)
library(Biostrings)
biomfile <- "data/affiliation_16S.biom"
frogs16S <- import_frogs(biomfile, taxMethod = "blast")
metadata <- read.table("data/metadata_16S.txt", row.names = 1, header = TRUE, sep = "\t", stringsAsFactors = FALSE)
phy_tree(frogs16S) <- read_tree("data/tree_16S.nwk")
sample_data(frogs16S) <- metadata
frogs16Sphyloseq-class experiment-level object
otu_table() OTU Table: [ 1250 taxa and 35 samples ]
sample_data() Sample Data: [ 35 samples by 3 sample variables ]
tax_table() Taxonomy Table: [ 1250 taxa by 7 taxonomic ranks ]
phy_tree() Phylogenetic Tree: [ 1250 tips and 1249 internal nodes ]fasta_file <- "data/asv_16S.fasta"
sequences_16S <- readDNAStringSet(fasta_file)
#taxa_names(frogs16S) <- unlist(as.character(sequences_16S))
clusters_in_frogs16S <- taxa_names(frogs16S)
matched_sequences <- sequences_16S[match(clusters_in_frogs16S, names(sequences_16S))]
# Check correspondance
if (any(is.na(matched_sequences))) {
stop("Some ASVs are not in the FASTA file! Please check...")
}
taxa_names(frogs16S) <- as.character(matched_sequences)Remove Mitochondria and Chloroplast
Let look at the proportion of Mitochondria and Chloroplast…
frogs16S_order <- tax_glom(frogs16S, taxrank = "Order") # Agglomerate at Order level to assess chloroplast proportion
# Calculate relative abundance
frogs16S_order.prop <- transform_sample_counts(frogs16S_order, function(x) x / sum(x) )
# Subset object to only Chloroplast
frogs16S_order.prop.chhloro <- subset_taxa(frogs16S_order.prop, Order == "Chloroplast")
tchloro <- otu_table(frogs16S_order.prop.chhloro) %>% as.data.frame()
rownames(tchloro) <- c("Chloroplast")
frogs16S_family <- tax_glom(frogs16S, taxrank = "Family")
frogs16S_family.prop <- transform_sample_counts(frogs16S_family, function(x) x / sum(x) )
frogs16S_family.prop.mito <- subset_taxa(frogs16S_family.prop, Family == "Mitochondria")
tmito <- otu_table(frogs16S_family.prop.mito) %>% as.data.frame()
rownames(tmito) <- c("Mitochondria")
library(reactable)
reactable(rbind(tchloro, tmito), defaultColDef = colDef(format = colFormat(digits = 2)))Then, I remove Mitochondria and Chloroplast of the dataset:
frogs16S <- frogs16S %>% subset_taxa(Order != "Chloroplast" & Family != "Mitochondria")
frogs16Sphyloseq-class experiment-level object
otu_table() OTU Table: [ 1088 taxa and 35 samples ]
sample_data() Sample Data: [ 35 samples by 3 sample variables ]
tax_table() Taxonomy Table: [ 1088 taxa by 7 taxonomic ranks ]
phy_tree() Phylogenetic Tree: [ 1088 tips and 1087 internal nodes ]Finally I save my phyloseq object in a RDS file:
saveRDS(frogs16S,file="html/frogs16S.rds")Affiliations
taxonomy_table <- as.data.frame(tax_table(frogs16S))
clusters <- taxa_names(frogs16S)
abundances <- as.data.frame(otu_table(frogs16S))
abundance_global <- rowSums(abundances)
result_tibble <- taxonomy_table %>%
rownames_to_column(var = "ASV") %>%
mutate(Abundance = abundance_global)
result_tibble <- as_tibble(result_tibble)
result_tibble %>% datatable()write.table(result_tibble, "html/16S_affiliations.txt", append=TRUE, quote = FALSE, row.names = FALSE, col.names = TRUE, sep = "\t" )This table is downloadable at the end of this document.
Compositions
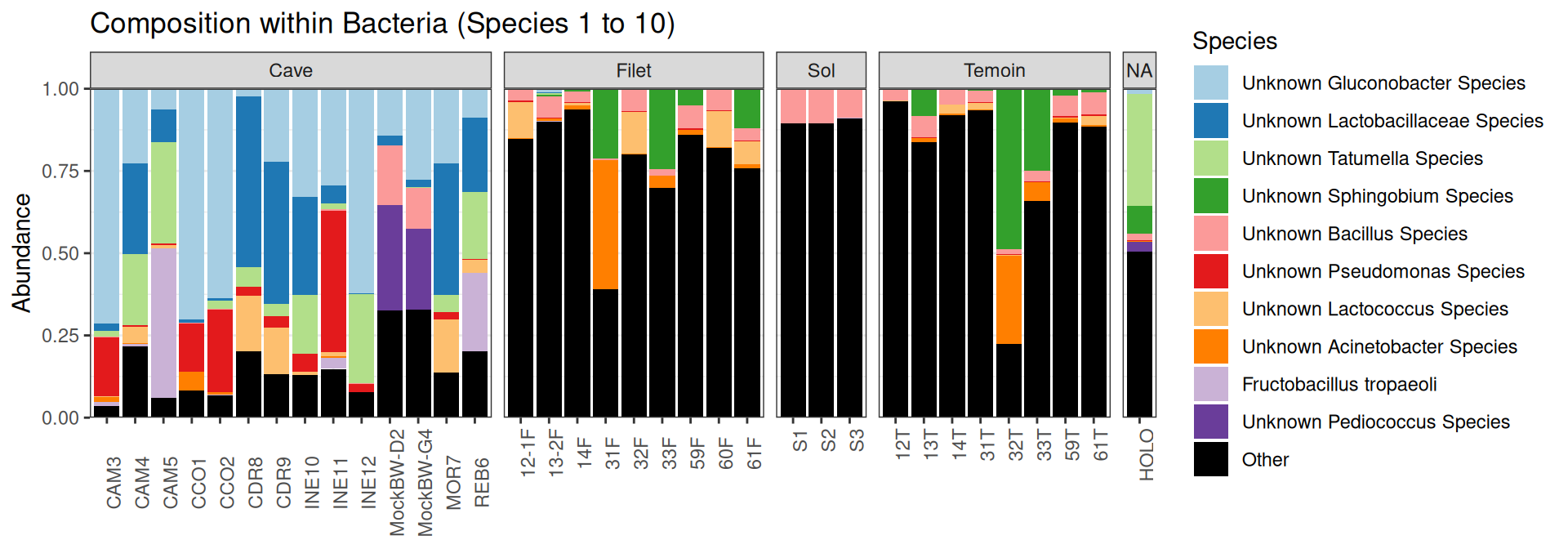
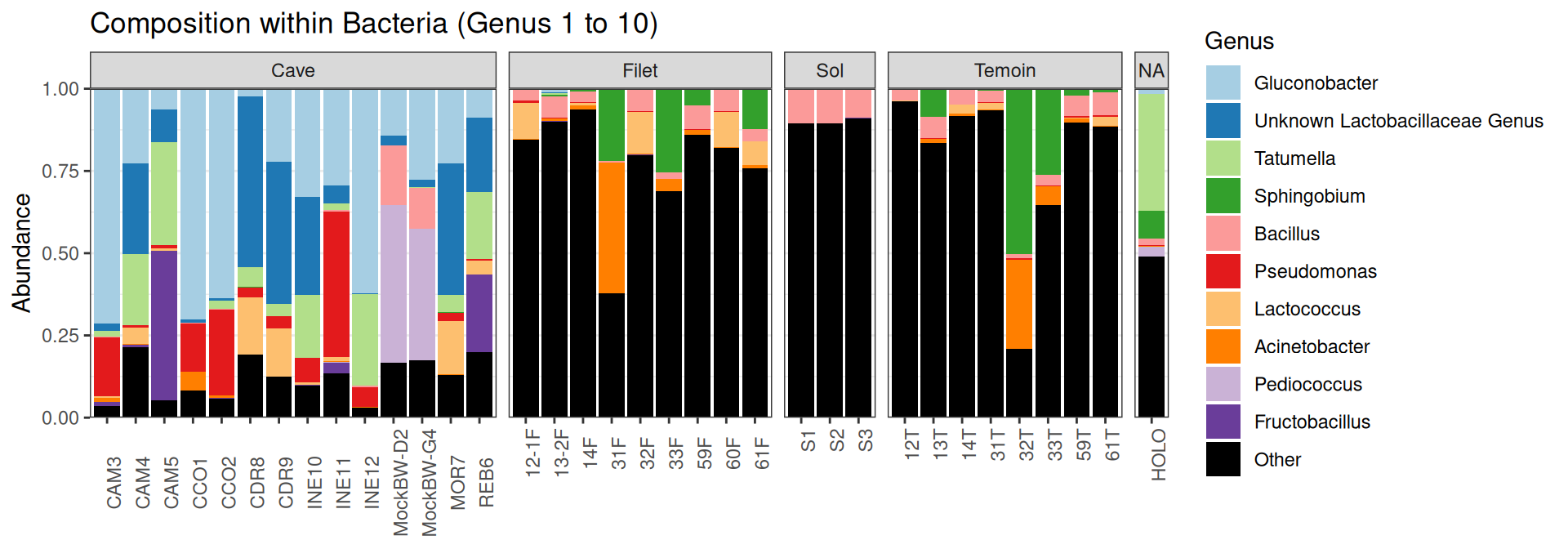
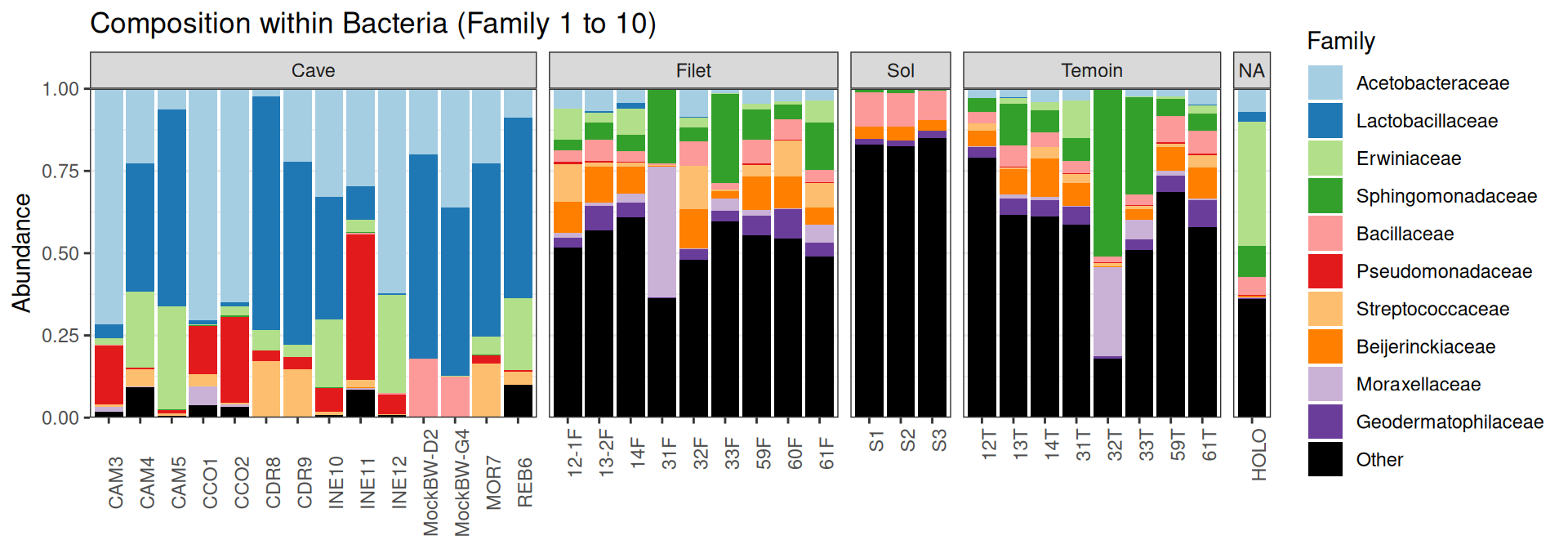
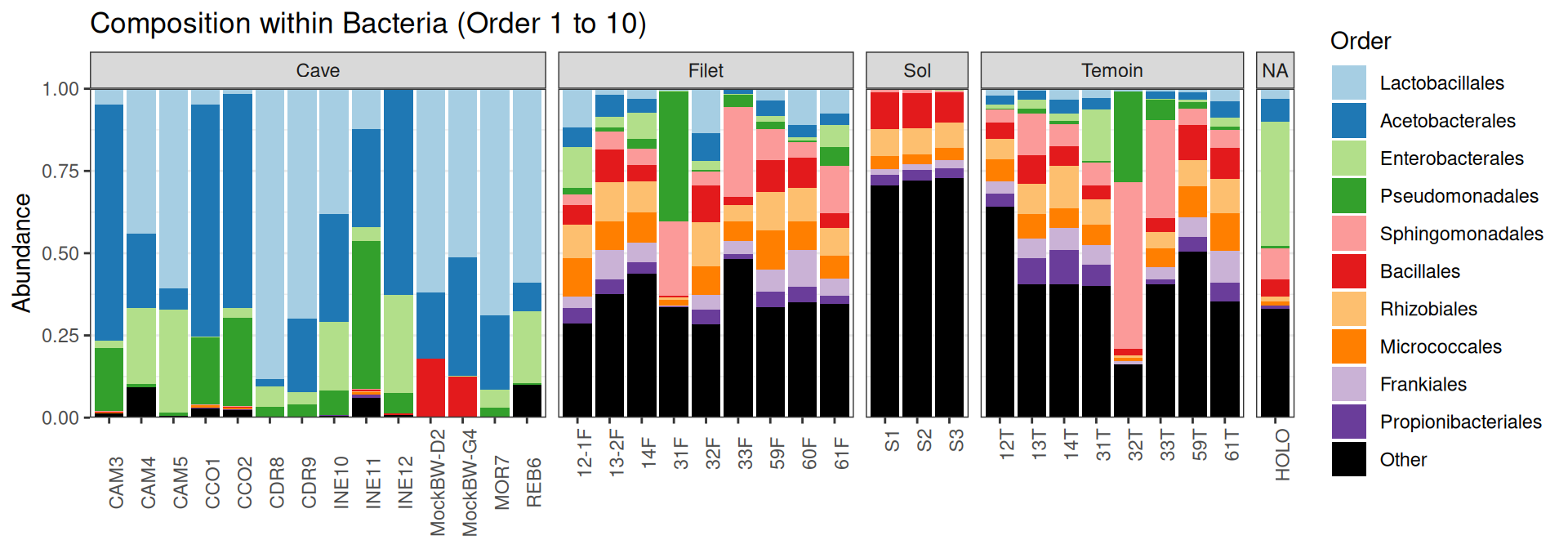
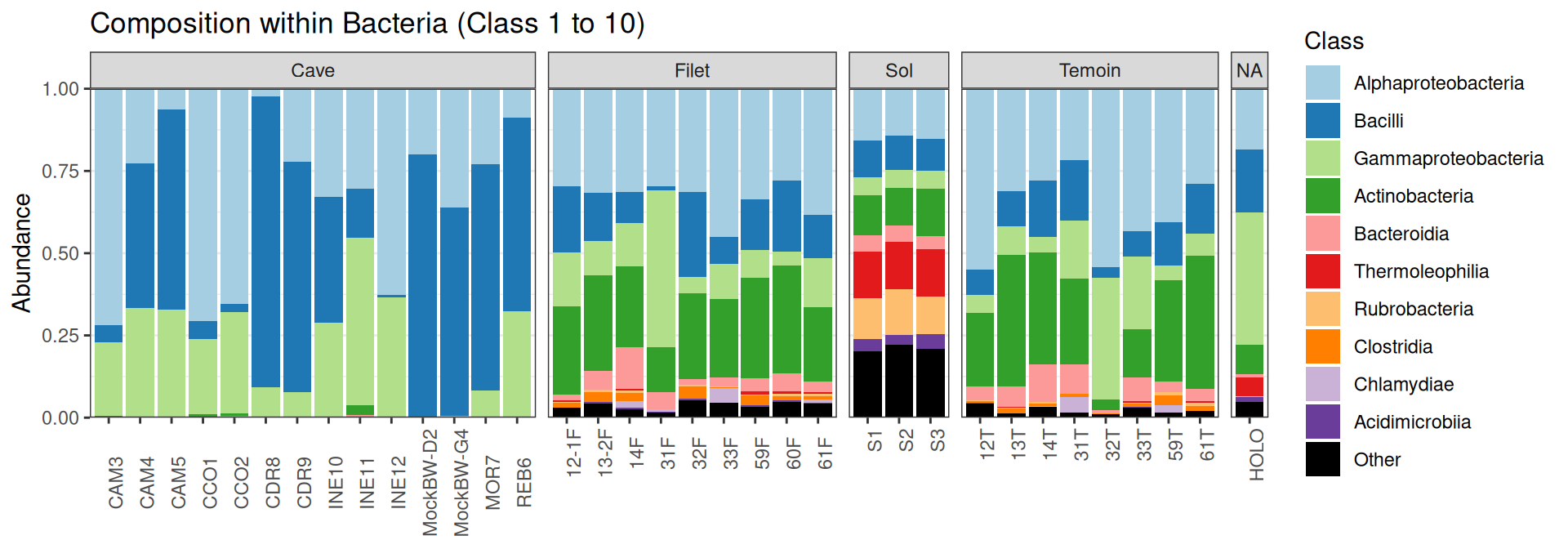
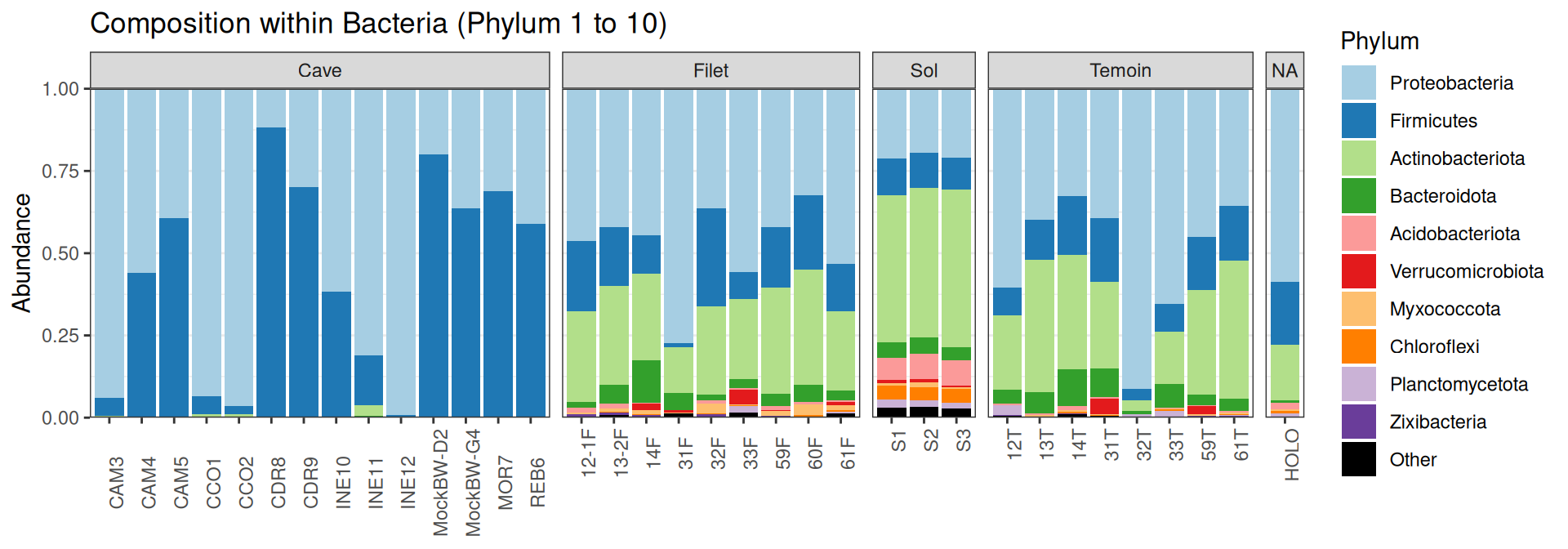
cat("\n")To create a phyloseq object, we need the BIOM file, the metadata file and eventually a tree file.
library(phyloseq)
library(phyloseq.extended)
biomfile <- "data/affiliation_ITS.biom"
frogsITS <- import_frogs(biomfile, taxMethod = "blast")
phy_tree(frogsITS) <- read_tree("data/tree_ITS.nwk")
metadata <- read.table("data/metadata_ITS.txt", row.names = 1, header = TRUE, sep = "\t", stringsAsFactors = FALSE)
sample_data(frogsITS) <- metadata
frogsITSphyloseq-class experiment-level object
otu_table() OTU Table: [ 649 taxa and 35 samples ]
sample_data() Sample Data: [ 35 samples by 3 sample variables ]
tax_table() Taxonomy Table: [ 649 taxa by 7 taxonomic ranks ]
phy_tree() Phylogenetic Tree: [ 649 tips and 648 internal nodes ]fasta_file <- "data/asv_ITS.fasta"
sequences_ITS <- readDNAStringSet(fasta_file)
clusters_in_frogsITS <- taxa_names(frogsITS)
matched_sequences <- sequences_ITS[match(clusters_in_frogsITS, names(sequences_ITS))]
# Check correspondance
if (any(is.na(matched_sequences))) {
stop("Some ASVs are not in the FASTA file! Please check...")
}
taxa_names(frogsITS) <- as.character(matched_sequences)Some affiliations have to be changed, due to differences in some taxonomies (i.e. Pichia affiliated as Pichiaceae family vs. Saccharomycetaceae family)
change_complete_taxo <- function(t, taxo, sequence){
taxolist <- unlist(strsplit(taxo, ";"))
if(sequence %in% rownames(t)){
t[sequence,"Kingdom"] <- taxolist[1]
t[sequence,"Phylum"] <- taxolist[2]
t[sequence,"Class"] <- taxolist[3]
t[sequence,"Order"] <- taxolist[4]
t[sequence,"Family"] <- taxolist[5]
t[sequence,"Genus"] <- taxolist[6]
t[sequence,"Species"] <- taxolist[7]
}
return(t)
}
t <- phyloseq::tax_table(frogsITS)
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Pichiaceae;Pichia;Pichia_kluyveri","GAAATGCGATACCTAGTGTGAATTGCAGCCATCGTGAATCATCGAGTTCTTGAACGCACATTGCGCCCCATGGTATTCCATGGGGCATGCCTGTCTGAGCGTCGTTTCCTTCTTGCGCAAGCAGAGTTGAGAACAGGCTATGCCTTTTTCGAAATGGAACGTCGTGGACGAAGTGAACTAAATTATTGGAACGCTTTGGCCGCCGAACTTTTAACTAAGCTCGACCTCAGATCAGGTAGGAATACCCGCTGAACTTAA")
phyloseq::tax_table(frogsITS) <- tAffiliations
taxonomy_table <- as.data.frame(tax_table(frogsITS))
clusters <- taxa_names(frogsITS)
abundances <- as.data.frame(otu_table(frogsITS))
abundance_global <- rowSums(abundances)
result_tibble <- taxonomy_table %>%
rownames_to_column(var = "ASV") %>%
mutate(Abundance = abundance_global)
result_tibble <- as_tibble(result_tibble)
result_tibble %>% datatable()Modifications of some affiliations
t <- phyloseq::tax_table(frogsITS)
change_complete_taxo <- function(t, taxo, sequence){
taxolist <- unlist(strsplit(taxo, ";"))
if(sequence %in% rownames(t)){
t[sequence,"Kingdom"] <- taxolist[1]
t[sequence,"Phylum"] <- taxolist[2]
t[sequence,"Class"] <- taxolist[3]
t[sequence,"Order"] <- taxolist[4]
t[sequence,"Family"] <- taxolist[5]
t[sequence,"Genus"] <- taxolist[6]
t[sequence,"Species"] <- taxolist[7]
}
return(t)
}
t <- change_complete_taxo(t,"Fungi;Basidiomycota;Agaricostilbomycetes;Agaricostilbales;Kondoaceae;Kondoa;Kondoa_aeria","GAAATGCGATACGTAATGTGAATTGCAGAACTCAGTGAATCATCGAATTTTTGAACGCACCTTGCGCTCTTTAGTTATTCTGAAGAGCATGCTTGTTTGAGTGTCGCGAACCTCTCAACCCCGCCCAAGGCTTAATTGACTTGCGGTTGTTTGGGCTTGGATCATGGTCTCTTGTCGTTACCTTCGGGTTTGGACTGGACTGAAATACAACAAGTTGAATGGTTTCCATTTGCAGCTTGACCTGATGTTGTAAACTACTCATCGGGGATGCAATGAAGCTAAAGGAACTTTAACAGAACCCCTTAAATGGTACCTAGTACATTTATGACCTCAAATCAAGTAGGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Basidiomycota;Agaricomycetes;Russulales;Peniophoraceae;Entomocorticium;Entomocorticium_sp","GAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACCTTGCGCCCCTTGGCATTCCGAGGGGCACGCCTGTTTGAGTGTCGTGAACTCCTCCACCCTCTACCTTTTTCGGAAGGCACTGGGCTGGGATTTGGGAGCTTGCGGGTCCCTGGCCGATCCGCTCTCCTTGAATACATTAGCGAAGCCCTTGCGGCCTTGGTGTGATAGTCATCTACGCCTTGGCTTAGCGAACATATGGGAATCGCTTCCAACCGTCTCGCAAGAGACAATCACTACCAACTTGACCTCAAATCAGGCGGGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Basidiomycota;Agaricomycetes;Thelephorales;Thelephoraceae;Tomentella;Tomentella_tedersooi","TAAATGTGACAATTAATGTGACTTGCAGAGTACGTGAATCATCAAGTATTTGAATGCACATTGCACTTTCTGTTCAAGAAAGTATACCTGTTTGAGTACCATATTCTTTTCCCTTTAATGGAAACTTGTGTCATCCGTTGCGCTTTTAGCGGCGACGGCGATACAAATTAAAGAGTCCAACTCGAGACGTAAAAACATTGCAAAATGATTGAACGTTGCGCGTTGCTTTACTACAACTTTTTTGAATTGATCTCAAATCAGGCAGGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Basidiomycota;Agaricomycetes;Thelephorales;Thelephoraceae;Tomentella;Tomentella_tedersooi","TAAATGTGACAATTAATGTGACTTGCAGAGTACGTGAATCATCAAGTATTTGAATGCACATTGCACTTTCTGTTCAAGAAAGTATACCTGTTTGAGTACCATATTCTTTTCCCTTTAATGGAAACTTGTGTCATCCGTTGCGCTTCTAGCGGCGACGGCGATACAAATTAAAGAGTCCAACTCGAGACGTAAAAACATTGCAAAATGATTGAACGTTGTGCGTTGCTTTACTACAACTTTTTTGAATTGATCTCAAATCAGGCAGGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Mortierellomycota;Mortierellomycetes;Mortierellales;Mortierellaceae;Mortierella;Mortierella_alpina","GAAATGCGATACGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCATATTGCGCTCTCTGGTATTCCGGAGAGCATGCTTGTTTGAGTATCAGTAAACACCTCAACTCCCTTTTCTTTTTTGAAATTGGAGCTGGACTTGAGTGATCCCAACGCTTTCTTCCAAGAAAGTGGCGGGTTGCTTGAAATGCAGGTGCAGCTGAACTTTTCTCTGAGCTATAAGCATATCTATTTAGTCTGCCTAAAAAACAGATTATTACCTTTGCTGCAGCTAACATAAAGGAGACTAGTTCTTGTGCTGACTGATGCAGGATTCACAGAGACAGCTTCGGCTGACTTTGTAAACTCGATCTCAAATCAAGTAAGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Sordariomycetes;Hypocreales;Nectriaceae;Fusarium;Fusarium_sp","GAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAAGCCCCCGGGTTTGGTGTTGGGGATCGGGCTGTACTCCAGCCCGGCCCCGAAATCTAGTGGCGGTCTCGCTGCAGCCTCCATTGCGTAGTAGCTAACACCTCGCAACTGGAACGCGGCGCGGCCAAGCCGTTAAACCCCCAACTTCTGAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Sordariomycetes;Trichosphaeriales;Trichosphaeriaceae;Nigrospora;Nigrospora_sphaerica","GAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCATTAGTATTCTAGTGGGCATGCCTGTTCGAGCGTCATTTCAACCCCTAAGCACAGCTTATTGTTGGGAACCTACGGCTTCGTAGTTCCTCAAAGACATTGGCGGAGTGGCAGTGGTCCTCTGAGCGTAGTAATCTTTTATCTCGCTTCTGTTAGGTGCTGCCCCCCCGGCCGTAAAACCCCCAATTTTTTCTGGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Eurotiomycetes;Chaetothyriales;Trichomeriaceae;Knufia;Knufia_sp","GAAATGCGATAAGTAATGCGAATTGCAGAATTTCCGTGAGTCATCGAATCTTTGAACGCACATTGCGCCCACTGGTATTCCGGTGGGCATGCCTGTTCGAGCGTCATTATCCTCCCTCAAACCCCGGGTTTGGTGTTGGACCGAAGTTGTGTGAACAACTGGTCTAAAAGACAATGACGGCGTCCGTGGGACCCTCGGTGCAACGAGCTTTTAGGAGCACGCGCCGAGTTGCAAGGACCTTCCGGGCCGGTCTCCTTTTACTATTTTACAAGGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Basidiomycota;Tremellomycetes;Tremellales;Bulleribasidiaceae;Vishniacozyma;Vishniacozyma_dimennae","GAAATGCGATAAGTAGTGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCCTCGGTATCCCGGGGGGCATGCCTGTTCGAGCGTCATTTCTACCACTCAAGCACAGCTTGGTATTGGGTGTCGTTGCTTTTCTAGCCAACGTGCCTTAAATGTAGACGGCAGCAAGCACCAGTTCCGAGCGTAGCAGAAAACTCGCTTTAGGGGCTTTGGGGCATTGCTAACCGCATAAGCTTTTTTATAAGTTTGACCTCGGATCAGGCAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Dothideomycetes;Cladosporiales;Cladosporiaceae;Cladosporium;Multi-affiliation","GAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCCCTGGTATTCCGGGGGGCATGCCTGTTCGAGCGTCATTTCACCACTCAAGCCTCGCTTGGTATTGGGCAACGCGGTCCGCCGCGTGCCTCAAATCGTCCGGCTGGGTCTTCTGTCCCCTAAGCGTTGTGGAAACTATTCGCTAAAGGGTGTCCGGGAGGCTACGCCGTAAAACAACCCCATTTCTAAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Dothideomycetes;Cladosporiales;Cladosporiaceae;Cladosporium;Multi-affiliation","GAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCCCTGGTATTCCGGGGGGCATGCCTGTTCGAGCGTCATTTCACCACTCAAGCCTCGCTTGGTATTGGGCAACGCGGTCCGCCGCGTGCCTCAAATCGTCCGGCTGGGTCTTCTGTCCCCTAAGCGTTGTGGAAACTATTCGCTAAAGGGTGTTCGGGAGGCTACGCCGTAAAACAACCCCATTTCTAAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Dothideomycetes;Cladosporiales;Cladosporiaceae;Cladosporium;Multi-affiliation","GAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCCTTGGTATTCCGAGGGGCATGCCTGTTCGAGCGTCATTTCACCACTCAAGCCTCGCTTGGTATTGGGCAACGCGGTCCGCCGCGTGCCTCAAATCGTCCGGCTGGGTCTTCTGTCCCCTAAGCGTTGTGGAAACTATTCGCTAAAGGGTGTTCGGGAGGCTACGCCGTAAAACAACCCCATTTCTAAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Dothideomycetes;Cladosporiales;Cladosporiaceae;Cladosporium;Multi-affiliation","GAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCTTTGGTATTCCAAAGGGCATGCCTGTTCGAGCGTCATTTCACCACTCAAGCCTCGCTTGGTATTGGGCAACGCGGTCCGCCGCGTGCCTCAAATCGTCCGGCTGGGTCTTCTGTCCCCTAAGCGTTGTGGAAACTATTCGCTAAAGGGTGTTCGGGAGGCTACGCCGTAAAACAACCCCATTTCTAAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Dothideomycetes;Cladosporiales;Cladosporiaceae;Cladosporium;Multi-affiliation","GAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCCTTGGTATTCCGGGGGGCATGCCTGTTCGAGCGTCATTTCACCACTCAAGCCTCGCTTGGTATTGGGCAACGCGGTCCGCCGCGTGCCTCAAATCGTCCGGCTGGGTCTTCTGTCCCCTAAGCGTTGTGGAAACTATTCGCTAAAGGGTGTTCGGGAGGCTACGCCGTAAAACAACCCCATTTCTAAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Dothideomycetes;Cladosporiales;Cladosporiaceae;Cladosporium;Multi-affiliation","GAAATGCGATAAGTAATGTGAATTGCAGAATTCCGTGAATCATCGAATCTTTGAACGCACATTGCGCCCCCTGGTATTCCGGGGGGCATGCCTGTTCGAGCGTCATTTCACCACTCAAGCCTCGCTTGGTATTGGGCAACGCGGTCCGCCGCGTGCCTCAAATCGTCCGGCTGGGTCTTCTGTCCCCTAAGCGTTGTGGAAACTATTCGCTAAAGGGTGTTCGGGAGGCTACGCCGTAAAACAACCCCATTTCTAAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Dothideomycetes;Cladosporiales;Cladosporiaceae;Cladosporium;Multi-affiliation","GAAATGCGATAACTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCCCTGGTATTCCGGGGGGCATGCCTGTTCGAGCGTCATTTCACCACTCAAGCCTCGCTTGGTATTGGGCAACGCGGTCCGCCGCGTGCCTCAAATCGTCCGGCTGGGTCTTCTGTCCCCTAAGCGTTGTGGAAACTATTCGCTAAAGGGTGTCCGGGAGGCTACGCCGTAAAACAACCCCATTTCTAAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Dothideomycetes;Cladosporiales;Cladosporiaceae;Cladosporium;Multi-affiliation","GAAATGCGATAACTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCCCTGGTATTCCGGGGGGCATGCCTGTTCGAGCGTCATTTCACCACTCAAGCCTCGCTTGGTATTGGGCAACGCGGTCCGCCGCGTGCCTCAAATCGTCCGGCTGGGTCTTCTGTCCCCTAAGCGTTGTGGAAACTATTCGCTAAAGGGTGTTCGGGAGGCTACGCCGTAAAACAACCCCATTTCTAAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Dothideomycetes;Cladosporiales;Cladosporiaceae;Cladosporium;Multi-affiliation","GAAATGCGATACGTAGTGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCCCTGGTATTCCGGGGGGCATGCCTGTTCGAGCGTCATTTCACCACTCAAGCCTCGCTTGGTATTGGGCAACGCGGTCCGCCGCGTGCCTCAAATCGACCGGCTGGGTCTTCTGTCCCCTAAGCGTTGTGGAAACTATTCGCTAAAGGGTGTTCGGGAGGCTACGCCGTAAAACAACCCCATTTCTAAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Dothideomycetes;Cladosporiales;Cladosporiaceae;Cladosporium;Multi-affiliation","GAAATGCGATACGTAGTGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCCCTGGTATTCCGGGGGGCATGCCTGTTCGAGCGTCATTTCACCACTCAAGCCTCGCTTGGTATTGGGCAACGCGGTCCGCCGCGTGCCTCAAATCGTCCGGCTGGGTCTTCTGTCCCCTAAGCGTTGTGGAAACTATTCGCTAAAGGGTGTTCGGGAGGCTACGCCGTAAAACAACCCCATTTCTAAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Dothideomycetes;Cladosporiales;Cladosporiaceae;Cladosporium;Multi-affiliation","GAAATGCGATAAGTAGTGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCCCTGGTATTCCGGGGGGCATGCCTGTTCGAGCGTCATTTCACCACTCAAGCCTCGCTTGGTATTGGGCAACGCGGTCCGCCGCGTGCCTCAAATCGTCCGGCTGGGTCTTCTGTCCCCTAAGCGTTGTGGAAACTATTCGCTAAAGGGTGTTCGGGAGGCTACGCCGTAAAACAACCCCATTTCTAAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Dothideomycetes;Pleosporales;Torulaceae;Torula;Torula_ligniperda","GAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCCTTGGTATTCCGAGGGGCATGCCTGTTCGAGCGTCATTTCAACCCCTCAAGCTTAGCTTGGTGTTGGGCTGCGCCAGCGTTGCTGGCGGGCCTTAAAATCAGTGGCGGTGCCGTTTGGGCTCCAAGCGTAGTAGCATCTCTCGCTCTGGAGACCCGGCGGTTGCTTGCCAGACAATCACTAAAAAAACAAAGGTTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Leotiomycetes;Helotiales;Sclerotiniaceae;Stromatinia;Multi-affiliation","GAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCCTTGGTATTCCGGGGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAAGCTCAGCTTGGTATTGGGCCTCCGCCGGTCACACGGCGGGCCTTAAAGTCAGTGGCGGCGCCGTTGGGTCCTGAACGTAGTAACATACATCTCGTTACAGGGTCCCCGCGTGCTTCTGCCATTAAACCCCCAATTTCTATGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Leotiomycetes;Helotiales;Ploettnerulaceae;Cadophora;Cadophora_luteo-olivacea","GAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCTCTGGTATTCCGGGGGGCATGCCTGTTCGAGCGTCATTATAACCACTCAAGCTCTCGCTTGGTATTGGGGTTCGCGGTTTCGCGGCTCCTAAAATCAGTGGCGGTGCCTGTCGGCTCTACGCGTAGTAATACTCCTCGCGTCTGGGTCCGGTAGGTCTACTTGCCAGCAACCCCCAATTTTTACAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Dothideomycetes;Pleosporales;Sporormiaceae;Preussia;Multi-affiliation","GAAATGCGATAAGTAGTGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCTTTGGTATTCCTTAGGGCATGCCTGTTCGAGCGTCATTTAAACCTTCAAGCTCAGCTTGGTGTTGGGTGACTGTCCGCTTCACTGCGGACTCGCCTCAAAATTATTGGCGGCCGGTACATTGGCTTCGAGCGCAGCAGAAACGCGAACTCGGGCCCGTCGTATTGGCTCCCAGAAGCTATCTTCACAATTTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Dothideomycetes;Pleosporales;Sporormiaceae;Sporormiella;Sporormiella_sp","GAAATGCGATAAGTAGTGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCTTTGGTATTCCTTAGGGCATGCCTGTTCGAGCGTCATTTAAACCTTCAAGCTAAGCTTGGTGTTGGGTGACTGTCCGCTTCACGGCGGACTCGCCTCAAAATTATTGGCGGCCGGTACATTGGCTTCGAGCGCAGCAGAAACGCGAACTCGGGCCCGTCGTATTGGCTCCCAGAAGCTATCTTCACAATTTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Dothideomycetes;Pleosporales;Lophiostomataceae;Lophiostoma;Lophiostoma_sp","GAAATGCGATAAGTAGTGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCTTTGGTATTCCTTAGGGCATGCCTGTTCGAGCGTCATTTACAAATTCAAGCTCAGCTTGGTGATGGGTGTCTGTCCCGCCTTTGCGTGTGGACTCGCCTCAAATGCAGTTGGCAGCTTGTTCCTCGGCTCTAAACGCAGCAGATTTGCGTCGAGCGTCGTGCGGACGGGCTCTCCAGTAAGCAAACCCCACAAATTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Dothideomycetes;Pleosporales;Cucurbitariaceae;Pyrenochaeta;Pyrenochaeta_sp","GAAATGCGATAAGTAGTGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCCTTGGTATTCCATGGGGCATGCCTGTTCGAGCGTCATTTGTACCCTCCAGCCCTGCTGGGTGTTGGGCGTTTGTTCCGCCGCGTGCGTGAACTCGCCTCAAATACATTGGCAGCCCGCCGTCCCGTGTGGGAGCGCAGCACATTTTGCGCTCTCCGCTGCAGACGGCGGCATCCACAAGTCTACACCTTTACGCTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Saccharomycodaceae;Hanseniaspora;Hanseniaspora_pseudoguilliermondii","GAATTGCGATAAGTAATGTGAATTGCAGATACTCGTGAATCATTGAATTTTTGAACGCACATTGCGCCCTTGAGCATTCTCAAGGGCATGCCTGTTTGAGCGTCATTTCCTTCTCAAAAGATAATTTATTATTTTTTGGTTGTGGGCGATACTCAGGGTTAGCTTGAAATTGGAGACTGTTTCAGTCTTTTTTAATTCAACACTTAGCTTCTTTGGAGACGCTGTTCTCGCTGTGATGTATTTATGGATTTATTCGTTTTACTTTACAAGGGAAATGGTAACGTACCTTAGGCAAAGGGTTGCTTTTAATATTCATCAAGTTTGACCTCAAATCAGGTAGGATTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Saccharomycetaceae;Pichia;Pichia_kluyveri","GAAATGCGATACCTAGTGTGAATTGCAGCCATCGTGAATCATCGAGTTCTTGAACGCACATTGCGCCCCATGGTATTCCATGGGGCATGCCTGTCTGAGCGTCGTTTCCTTCTTGCGCAAGCAGAGTTGAGAACAGGCTATGCCTTTTTCGAAATGGAACGTCGTGGACGAAGTGAACTAAACTTTTTAGCCCGCTTTGCCGGCCGAACTTTTTACTAAGCTCGACCTCAGATCAGGTAGGAATACCCGCTGAACTAAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Metschnikowiaceae;Metschnikowia;Metschnikowia_sp","GAATTGCGATAAGTAATGTGAATTGCAGATACTCGTGAATCATTGAATTTTTGAACGCACATTGCGCCCCGGGGTATTCCCCAGGGCATGCGTGGGTGAGCGATATTTACTCTCAAACCTCCGGTTTGGTCCTGCTTCGGCCTAATATCGACGGCGCTAGAATAAGTTTTAGCCCCAGCCTTTTTCCTCACCCTCGTAAGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Metschnikowiaceae;Metschnikowia;Metschnikowia_pulcherrima","GAATTGCGATACGTAATATGACTTGCAGACGTGAATCATTGAATTTTTGAACGCACATTGCGCCCCGGGGTATTCCCCAGGGCATGCGTGGGTGAGCGATATTTACTCTCAAACCTCCGGTTTGGTCCTGCTTCGGCCTAATATCAACGGCGCTAGAATAAGTTTTAGCCCCAGCCTTTTTCCTCACCCTCGTAAGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Metschnikowiaceae;Metschnikowia;Metschnikowia_pulcherrima","GAATTGCGATACGTAATATGACTTGCAGACGTGAATCATTGAATCTTTGAACGCACATTGCGCCCCGGGGTATTCCCCAGGGCATGCGTGGGTGAGCGATATTTACTCTCAAACCTCCGGTTTGGTCCTGCTTCGGCCTAATATCAACGGCGCTAGAATAAGTTTTAGCCCCAGTCTTTTTCCTCACCCTCGTAAGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Metschnikowiaceae;Metschnikowia;Metschnikowia_pulcherrima","GAATTGCGATACGTAATATGACTTGCAGACGTGAATCATTGAATCTTTGAACGCACATTGCGCCCCGGGGTATTCCCCAGGGCATGCGTGGGTGAGCGATATTTACTCTCAAACCTCCGGTTTGGTCCTGCTTCGGCCTAATATCAACGGCGCTAGAATAAGTTTTAGCCCCATCCTTTTTCCTCACCCTCGTAAGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Metschnikowiaceae;Metschnikowia;Metschnikowia_pulcherrima","GAATTGCGATACGTAATATGACTTGCAGACGTGAATCATTGAATCTTTGAACGCACATTGCGCCCCGGGGTATTCCCCAGGGCATGCGTGGGTGAGCGATATTTACTCTCAAACCTCCGGTTAGGTCCTGCTTCGGCCTAATATCAACGGCGCTAGAATAAGTTTTAGCCCCATTCTTTTTCCTCACCCTCGTAAGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Metschnikowiaceae;Metschnikowia;Metschnikowia_pulcherrima","CGACGAGCATAACAATAAGCGGAGGAACCCNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNTGGCATCGATGAAGAACGCAGCGAATTGCGATACGTAATATGACTTGCAGACGTGAATCATTGAATCTTTGAACGCACATTGCGCCCCGGGGTATTCCCCAGGGCATGCGTGGGTGAGCGATATTTACTCTCAAACCTCCGGTTTGGTCCTGCTTCGGCCTAATATCAACGGCGCTAGAATAAGTTTTAGCCCCATTCTTTTTCCTCACCCTCGTAAGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Metschnikowiaceae;Metschnikowia;Metschnikowia_pulcherrima","GAATTGCGATACGTAATATGACTTGCAGACGTGAATCATTGAATCTTTGAACGCACATTGCGCCCCGGGGTATTCCCCAGGGCATGCGTGGGTGAGCGATATTTACTCTCAAACCTCCGGTTTGGTCCTGCTTCGGCATAATATCAACGGCGCTAGAATAAGTTTTAGCCCCATTCTTCTTCCTCACCCTCGTAAGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Metschnikowiaceae;Metschnikowia;Metschnikowia_pulcherrima","GAATTGCGATACGTAATATGACTTGCAGACGTGAATCATTGAGTCTTTGAACGCACATTGCGCCCCGGGGTATTCCCCAGGGCATGCGTGGGTGAGCGATATTTACTCTCAAACCTCCGGTTTGGTCCTGCTTCGGCATAATATCAACGGCGCTAGAATAAGTTTTAGCCCCATTCTTCTTCCTCACCCTCGTAAGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Metschnikowiaceae;Metschnikowia;Metschnikowia_pulcherrima","GAATTGCGATACGTAATATGACTTGCAGACGTGAATCATTGAATTTTTGAACGCACATTGCGCCCCGGGGTATTCCCCAGGGCATGCGTGGGTGAGCGATATTTACTCTCAAACCTCCGGTTTGGTCCTGCTTCGGCATAATATCAACGGCGCTAGAATAAGTTTTAGCCCCATTCTTCTTCCTCACCCTCGTAAGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Metschnikowiaceae;Metschnikowia;Metschnikowia_pulcherrima","GAATTGCGATACGTAATATGACTTGCAGACGTGAATCATTGAGTCTTTGAACGCACATTGCGCCCCGGGGTATTCCCCAGGGCATGCGTGGGTGAGCGATATTTACTCTCAAACCTCCGGTTTGGTCCTGCTTCGGCCTAATATCAACGGCGCTAGAATAAGTTTTAGCCCCATTCTTCTTCCTCACCCTCGTAAGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Metschnikowiaceae;Metschnikowia;Metschnikowia_pulcherrima","GAATTGCGATACGTAATATGACTTGCAGACGTGAATCATTGAATCTTTGAACGCACATTGCGCCCCGGGGTATTCCCCAGGGCATGCGTGGGTGAGCGATATTTACTCTCAAACCTCCGGTTTGGTCCTGCTTCGGCCTAATATCAACGGCGCTAGAATAAGTTTTAGCCCCATTCTTCTTCCTCACCCTCGTAAGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Metschnikowiaceae;Metschnikowia;Metschnikowia_pulcherrima","GAATTGCGATACGTAATATGACTTGCAGACGTGAATCATTGAATCTTTGAACGCACATTGCGCCCCGGGGTATTCCCCAGGGCATGCGTGGGTGAGCGATATTTACTCTCAAGCCTCCGGTTTGGTCCTGCTTCGGCCTAATATCAACGGCGCTAGAATAAGTTTTAGCCCCATTCTTTTTCCTCACCCTCGTAAGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Metschnikowiaceae;Metschnikowia;Metschnikowia_pulcherrima","GAATTGCGATACGTAATATGACTTGCAGACGTGAATCATTGAATCTTTGAACGCACATTGCGCCCCGGGGTATTCCCCAGGGCATGCGTGGGTGAGCGATATTTACTCTCAAACCTCCGGTTTGGTCCTGCTTCGGCCTAATATCAACGGCGCTAGAATAAGTTTTAGCCCCATTCTTTTTCCTCACCCTCGTAAGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Metschnikowiaceae;Metschnikowia;Metschnikowia_pulcherrima","GAATTGCGATACGTAATATGACTTGCAGACGTGAATCATTGAGTCTTTGAACGCACATTGCGCCCCGGGGTATTCCCCAGGGCATGCGTGGGTGAGCGATATTTACTCTCAAACCTCCGGTTTGGTCCTGCTTCGGCCTAATATCAACGGCGCTAGAATAAGTTTTAGCCCCAGCCTTTTTCCTCACCCTCGTAAGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Metschnikowiaceae;Metschnikowia;Metschnikowia_pulcherrima","GAATTGCGATACGTAATATGACTTGCAGACGTGAATCATTGAATCTTTGAACGCACATTGCGCCCCGGGGTATTCCCCAGGGCATGCGTGGGTGAGCGATATTTACTCTCAAACCTCCGGTTTGGTCCTGCTTCGGCCTAATATCAACGGCGCTAGAATAAGTTTTAGCCCCAGCCTTTTTCCTCACCCTCGTAAGACTACCCGCTGAACTTAA")
t <- change_complete_taxo(t,"Fungi;Ascomycota;Saccharomycetes;Saccharomycetales;Metschnikowiaceae;Metschnikowia;Metschnikowia_pulcherrima","GAATTGCGATACGTAATATGACTTGCAGACGTGAATCATTGAATCTTTGAACGCACATTGCGCCCCGGGGTATTCCCCAGGGCATGCGTGGGTGAGCGATATTTACTCTCAAACCTCTGGTTTGGTCCTGCTTCGGCCTAATATCAACGGCGCTAGAATAAGTTTTAGCCCCAGCCTTTTTCCTCACCCTCGTAAGACTACCCGCTGAACTTAA")
phyloseq::tax_table(frogsITS) <- t
saveRDS(frogsITS,file="html/frogsITS.rds")taxonomy_table <- as.data.frame(tax_table(frogsITS))
clusters <- taxa_names(frogsITS)
abundances <- as.data.frame(otu_table(frogsITS))
abundance_global <- rowSums(abundances)
result_tibble <- taxonomy_table %>%
rownames_to_column(var = "ASV") %>%
mutate(Abundance = abundance_global)
result_tibble <- as_tibble(result_tibble)
result_tibble %>% datatable()write.table(result_tibble, "html/ITS_affiliations_mod2.txt", append=TRUE, quote = FALSE, row.names = FALSE, col.names = TRUE, sep = "\t" )This table is downloadable at the end of this document.
Compositions
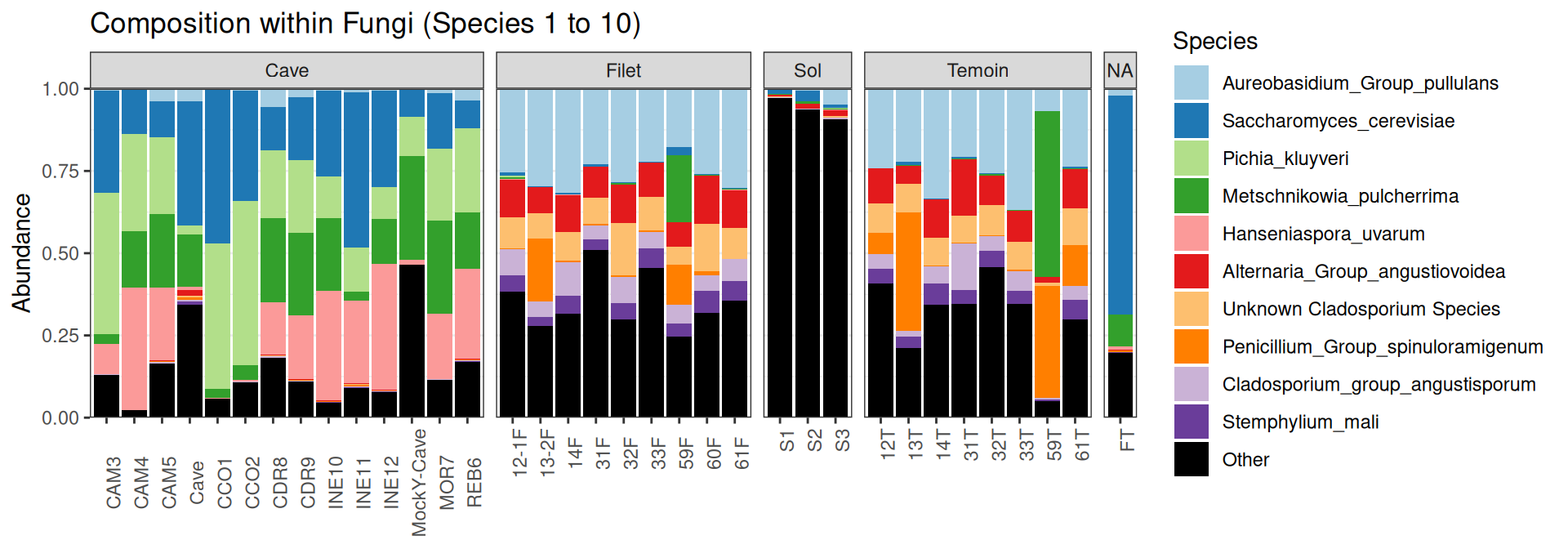
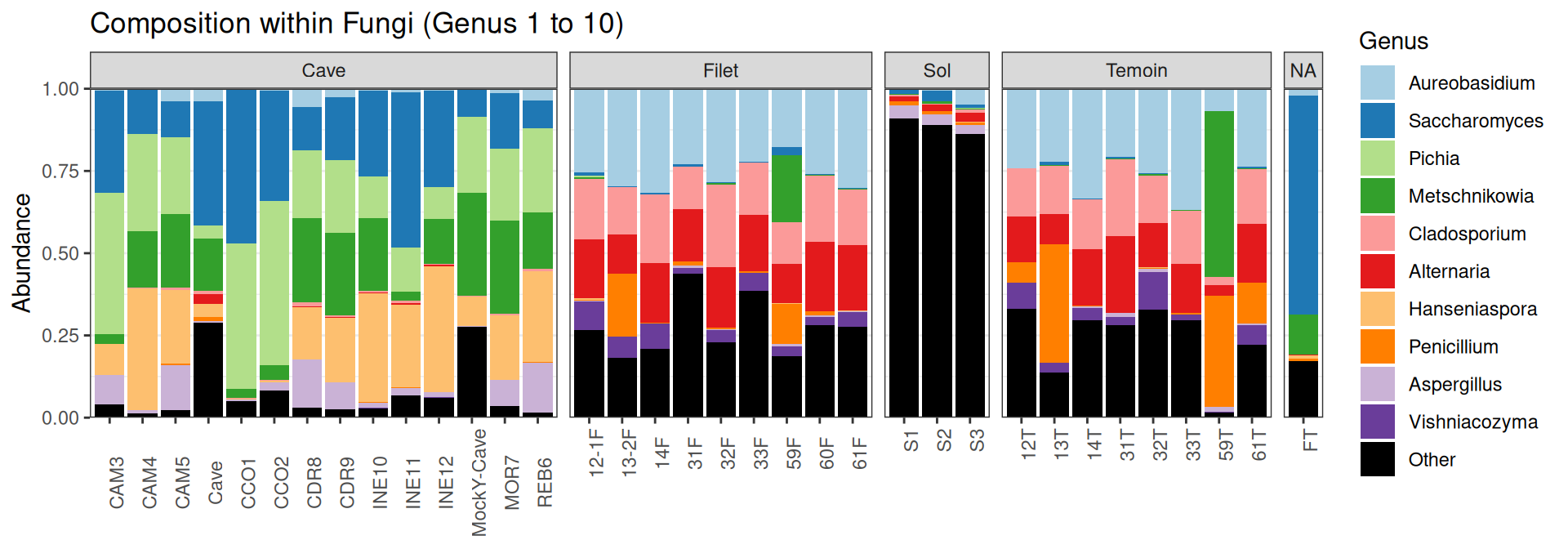
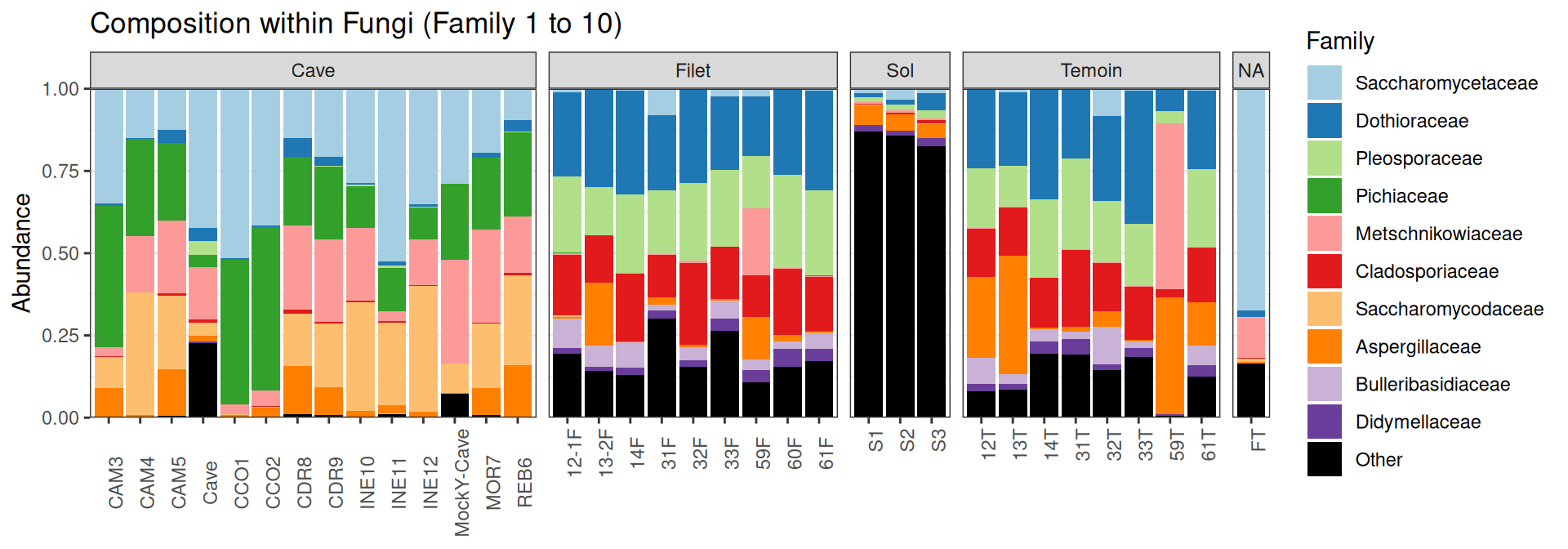
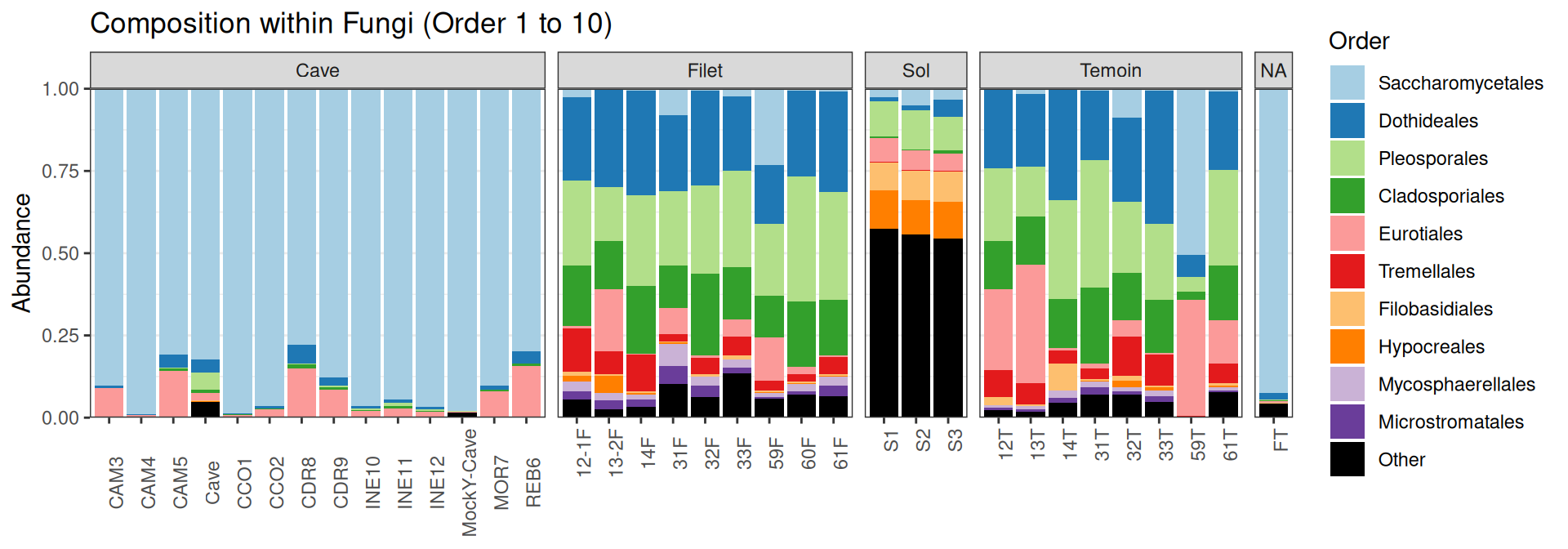
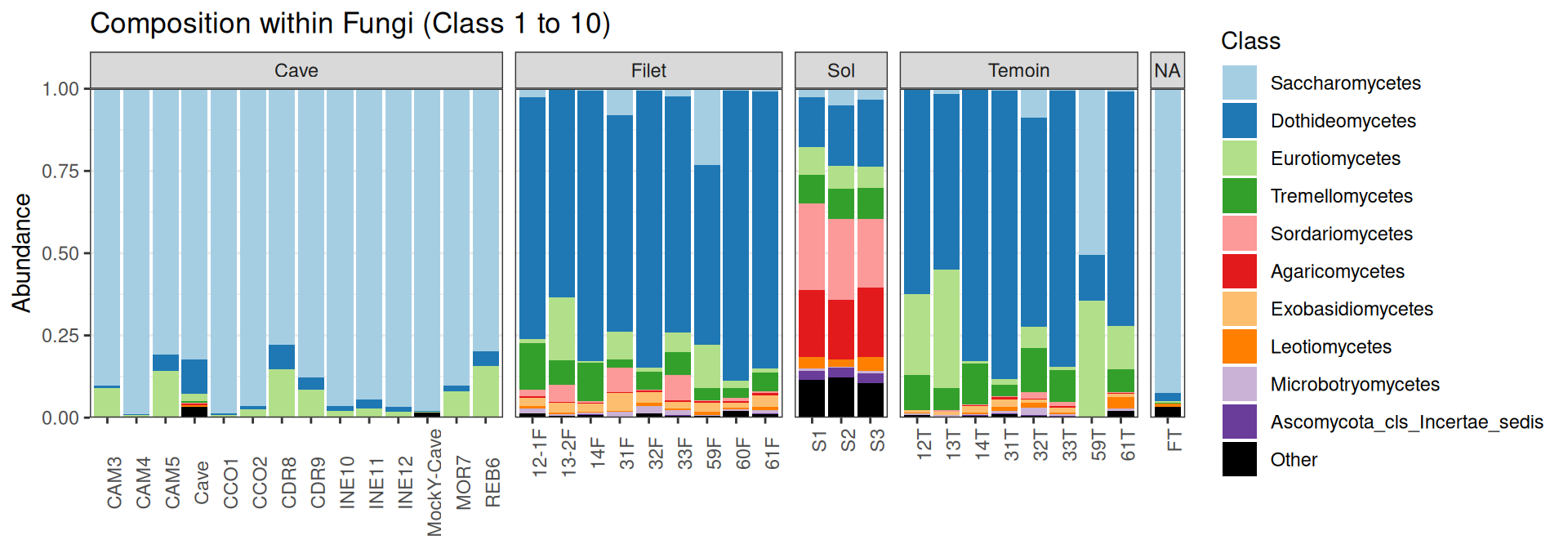
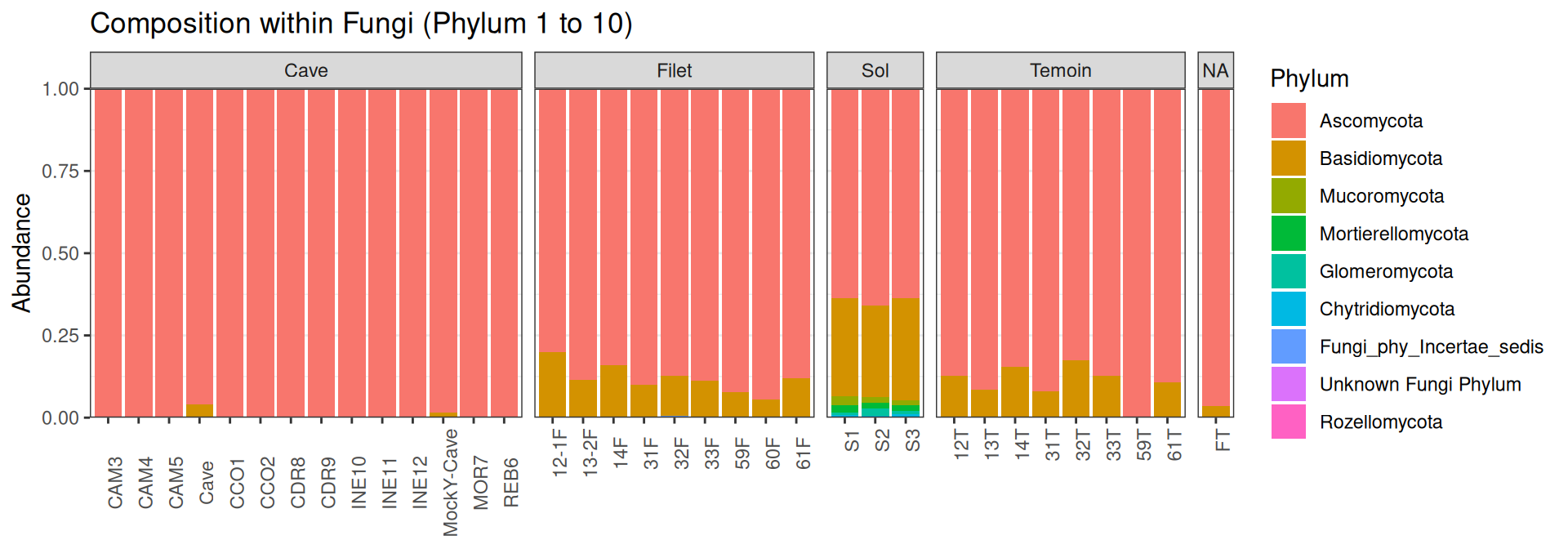
cat("\n")Downloads
The files can be used with Easy16S for an easy exploration.
- 16S rds file
- 16S affiliations text file
- ITS rds file
- ITS affiliations text file (old)
- ITS affiliations text file (after modifications)
- 16S TSV abundance file from original BIOM
- 16S TSV multihits from original BIOM
- ITS TSV abundance file from original BIOM
- ITS TSV multihits from original BIOM
You can add a column names Jean-Luc in the affiliation files to allow me a manual correction, the 7 ranks separated by a ; The abundance and multi-affiliation files can help you to choose/specify one particular affiliation. The join has to be made with the ASV sequence.
References
1. Shen W, Le S, Li Y, Hu F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/q file manipulation. PloS one. 2016;11:e0163962.
2. Andrews S. FastQC a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
3. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–8.
4. Escudié F, Auer L, Bernard M, Mariadassou M, Cauquil L, Vidal K, et al. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics. 2018;34:1287–94. doi:10.1093/bioinformatics/btx791.
5. Bernard M, Rué O, Mariadassou M, Pascal G. FROGS: a powerful tool to analyse the diversity of fungi with special management of internal transcribed spacers. Briefings in Bioinformatics. 2021;22. doi:10.1093/bib/bbab318.
6. Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from illumina amplicon data. Nature methods. 2016;13:581.
7. Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: A fast and accurate illumina paired-end reAd mergeR. Bioinformatics. 2013;30:614–20.
8. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet journal. 2011;17:10–2.
9. McMurdie PJ, Holmes S. Phyloseq: An r package for reproducible interactive analysis and graphics of microbiome census data. PloS one. 2013;8:e61217.
Reuse
This document will not be accessible without prior agreement of the partners
