
meta <- vroom::vroom("html/samples.tsv", delim = "\t", col_types = "fffff")
DT::datatable(meta)CAVIAR2
Whole genome analysis of Enterococcus cecorum
closed
collaboration
Note
This document is a report of the analyses performed. You will find all the code used to analyze these data. The version of the tools (maybe in code chunks) and their references are indicated, for questions of reproducibility.
Aim of the project
The aim of this project is to reuse CAVIAR workflow to analyze a new set of Enterococcus cecorum strains.
Patners
- Cédric Midoux - Migale bioinformatics facility - BioInfomics - INRAE
- Valentin Loux - Migale bioinformatics facility - BioInfomics - INRAE
- Pascale Serror - MICALIS - INRAE
Deliverables
Deliverables agreed at the preliminary meeting (Table 1).
| Definition | |
|---|---|
| 1 | Assembled genomes |
| 2 | Annotated genomes |
| 3 | HTML reports |
Data management
Important
All data is managed by the migale facility for the duration of the project. Once the project is over, the Migale facility does not keep your data. We will provide you with the raw data and associated metadata that will be deposited on public repositories before the results are used. We can guide you in the submission process. We will then decide which files to keep, knowing that this report will also be provided to you and that the analyses can be replayed if needed.
Raw data
The raw data is provided by the partner in directory front.migale.inrae.fr:/work_projet/cecotype/2022-CAVIAR-SEQUENCING/RAW and front.migale.inrae.fr:/work_projet/cecotype/2023-CAVIAR-SEQUENCING. It contains 59 shotgun sequencings of Enterococcus cecorum strains. The partner also provided a metadata table for the correspondence between strain name, library and experimental metadata.
Workflow
We reused a snakemake
This snakemake workflow aims to assemble Enterococcus cecorum genomes from raw reads and published reference data.

Config
The main parameters are specified in the config/config.yaml file.
They include :
| Variable | Definition | Default |
|---|---|---|
samples |
Table of strain metadata with path and experimental data. | /work_home/cmidoux/GIT/wf_caviar/config/samples.tsv |
raw_data |
Data path | /work_projet/cecotype |
workdir |
Results path | /work_projet/cecotype/caviar2-results |
subsample |
Size of sub-samples for easy assembly | 750000 |
reference |
Reference genome path, used by riboSeed |
/work_projet/cecotype/REF/NCTC12421/NCTC12421.fasta |
k_spades |
Size of k-mers used by spades |
21,33,55,77 |
genus |
Genus used by prokka for annotation |
Enterococcus |
species |
Species used by prokka for annotation |
cecorum |
proteins |
Gene catalogue path used by prokka for annotation |
/work_projet/cecotype/NANOPORE_ASSEMBLY-2020/Ref/Refseq/Enterococcus/proteins.faa |
eggnog_db |
eggNOG database path | /db/outils/eggnog-mapper/ |
kaiju_db |
Kaiju database path | /db/outils/kaiju-2021-03/nr_euk/ |
gene_detect |
Specific gene for BLAST detection | /work_home/cmidoux/GIT/wf_caviar/config/DQL78_RS09735.ffn |
Outputs
report/multiqc.html: MULTIQC report with :- FASTQC raw data quality report
- FASTP trimming report
- QUAST assembly report
- PROKKA annotation report
results/kaiju/krona.html&results/kaiju/kaiju.tsv: Raw data taxonomic annotation.results/ribo/ribo.tsv: riboSeed assembly metrics.results/assembly/{sample}/contigs.fasta: Sample assembly afterfastp,seqtk_subsample,riboSeedandspades.results/quast/report.html: Genome assembly evaluation.results/checkm/results.tsv: Assessment of genome quality (completeness and contamination) byCheckM.results/drep/: Clustering of assembled genomes.results/annot/prokka/{sample}/{sample}.gbk: Contig annotation byprokka.results/annot/eggnog/{sample}.emapper.hits: Functional annotation byeggNOG.results/gene_detect/{sample}.txt: Gene-specific alignment metrics.
Results & Notes
FASTQC
FASTQC
Note
The quality control (phread score, lenght, %GC) is good enough to go further.
Warning
Quality, adaptaer content and insert size of strain VE19254 [REMOVED] is worse than other sample.
Kaiju
Raw data are taxonomically annotated with kaiju nr_euk
Results are available in HTML report.
We made a representation at the “species” level.
vroom::vroom("html/kaiju.tsv", delim = "\t", col_types = "fddif") |>
mutate(strain = as_factor(stringr::str_split_i(file, pattern = "/", 3)), .before = 1, .keep = "unused") |>
mutate(taxon_name = fct_reorder(taxon_name, percent, .desc = TRUE, .na_rm = FALSE)) |>
left_join(meta, by = join_by(strain)) |>
ggplot(aes(fill = taxon_name, y = reads, x = strain)) +
geom_bar(position = "fill", stat = "identity") +
scale_fill_brewer(palette = "Set1", label = ~ stringr::str_wrap(.x, width = 50)) +
facet_grid(cols = vars(origin), scales = "free_x", space = "free") +
theme(axis.text.x = element_text(angle = 90, vjust = 0.5, hjust = 1)) +
theme(legend.position = "bottom") +
guides(fill = guide_legend(ncol = 2))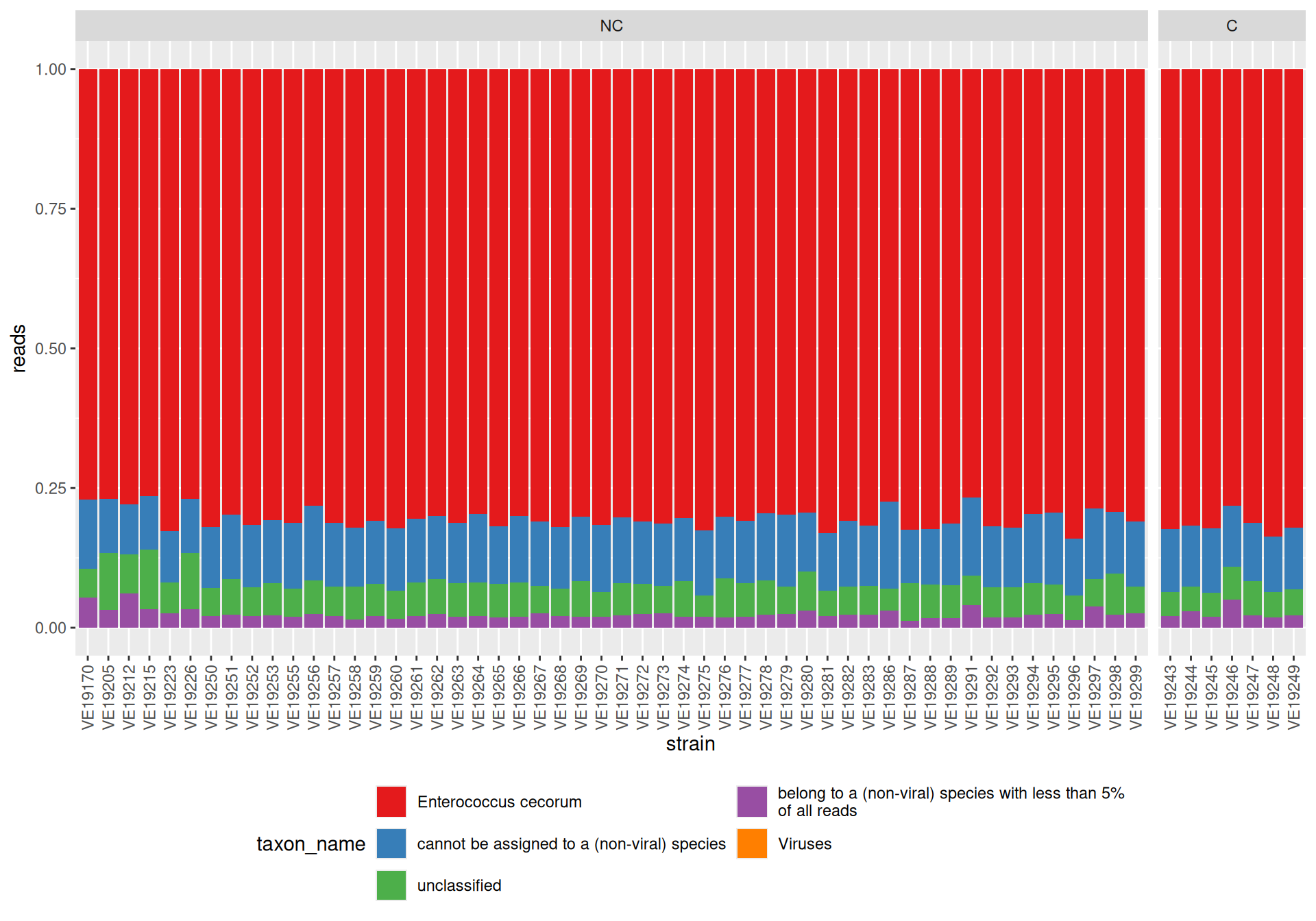
kaiju/nr_euk
Note
In contrast with CAVIAR1, no contamination has been observed.
riboSeed
riboSeed
read_tsv("html/ribo.tsv", col_types = "cffiiidi") |>
mutate(strain = stringr::str_split_i(file, pattern = "/", 3), .before = 1, .keep = "unused") |>
left_join(meta, by = join_by(strain)) |>
DT::datatable()
Important
riboSeed failed to produce contigs for strain VE19280.
SPAdes
We used SPAdes
Assembly results are available in QUAST report.
Important
Like for quality control, assembly look bad for strain VE19254 [REMOVED].
CheckM
We evaluate the robustness of assemblies with CheckM
checkm <- vroom::vroom("html/checkm.tsv", col_types = "ffiiiiiiiiiddd", .name_repair = snakecase::to_snake_case) |>
left_join(meta, by=join_by(bin_id==strain))
DT::datatable(checkm)Robustness estimation of genome
p <- ggplot(checkm, aes(x = completeness, y = contamination , col = strain_heterogeneity, shape = origin)) +
ggiraph::geom_point_interactive(aes(tooltip = bin_id)) +
expand_limits(x = c(0, 100), y = c(0, 100)) +
theme(legend.position = "bottom")
ggiraph::girafe(ggobj = p)Robustness estimation of genome
We also used dRep
read_csv("html/dRep_Cdb.csv", col_types = "cfdfff") |>
left_join(meta, by=join_by(genome==strain)) |>
DT::datatable()Primary clustering dendrogram and Secondary clustering dendrogram are available.
Note
MASH clustering build only one big cluster with 13 sub-clusters.
Details can be found in the table and figures.
PROKKA & eggNOG
Finally, we annotated contigs with pokka
Annotated data are available on results/annot/prokka/{sample}/{sample}.gbk following analysis.
After discussion, annotation with bakta results/annot/bakta/{sample}/{sample}.gbff.
References
1. Köster J, Rahmann S. Snakemake—a scalable bioinformatics workflow engine. Bioinformatics. 2012;28:2520–2.
2. Andrews S. FastQC a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
3. Menzel P, Ng KL, Krogh A. Fast and sensitive taxonomic classification for metagenomics with kaiju. Nature communications. 2016;7:11257.
4. Waters NR, Abram F, Brennan F, Holmes A, Pritchard L. riboSeed: Leveraging prokaryotic genomic architecture to assemble across ribosomal regions. Nucleic Acids Research. 2018;46:e68–8. doi:10.1093/nar/gky212.
5. Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology. 2012;19:455–77. doi:10.1089/cmb.2012.0021.
6. Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Research. 2015;25:1043–55. doi:10.1101/gr.186072.114.
7. Olm MR, Brown CT, Brooks B, Banfield JF. dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. The ISME Journal. 2017;11:2864–8. doi:10.1038/ismej.2017.126.
8. Seemann T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. doi:10.1093/bioinformatics/btu153.
9. Cantalapiedra CP, Hernández-Plaza A, Letunic I, Bork P, Huerta-Cepas J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Molecular Biology and Evolution. 2021;38:5825–9. doi:10.1093/molbev/msab293.
10. Schwengers O, Jelonek L, Dieckmann MA, Beyvers S, Blom J, Goesmann A. Bakta: Rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microbial Genomics. 2021;7. doi:https://doi.org/10.1099/mgen.0.000685.
Reuse
This document will not be accessible without prior agreement of the partners
