
Show the code
conda activate seqkit-2.0.0
seqkit stat Galaxy13-fna.fasta
file format type num_seqs sum_len min_len avg_len max_len
Galaxy13-fna.fasta FASTA DNA 449 3,116,159 227 6,940.2 805,683Genome analysis strategy to predict the proteolytic capacity of the strain Lacticaseibacillus casei CNCM I-5663
This document is a report of the analyses performed. You will find all the code used to analyze these data. The version of the tools (maybe in code chunks) and their references are indicated, for questions of reproducibility.
At Carembouche, we are developing a lactobacillus with probiotic properties by adding it to an oral nutritional supplement for malnourished people. As part of a collaboration with the STLO unit (INRAE, Rennes), we are seeking to evaluate the proteolytic capacities of our strain. We would also like to evaluate fibrolytic capacities. The genome of Lacticaseibacillus casei CNCM I-5663 has been sequenced by illumina.
Deliverables agreed at the preliminary meeting (Table 1).
| Definition | Status | |
|---|---|---|
| 1 | HTML report | ✔️ |
| 2 | Assembly (FASTA) | ✔️ |
| 3 | Annotation (GFF) | ✔️ |
All data is managed by the migale facility for the duration of the project. Once the project is over, the Migale facility does not keep your data. We will provide you with the raw data and associated metadata that will be deposited on public repositories before the results are used. We can guide you in the submission process. We will then decide which files to keep, knowing that this report will also be provided to you and that the analyses can be replayed if needed.
Genome assembly and prokka annotation were deposited on the front server and a copy was sent to the abaca server.
conda activate seqkit-2.0.0
seqkit stat Galaxy13-fna.fasta
file format type num_seqs sum_len min_len avg_len max_len
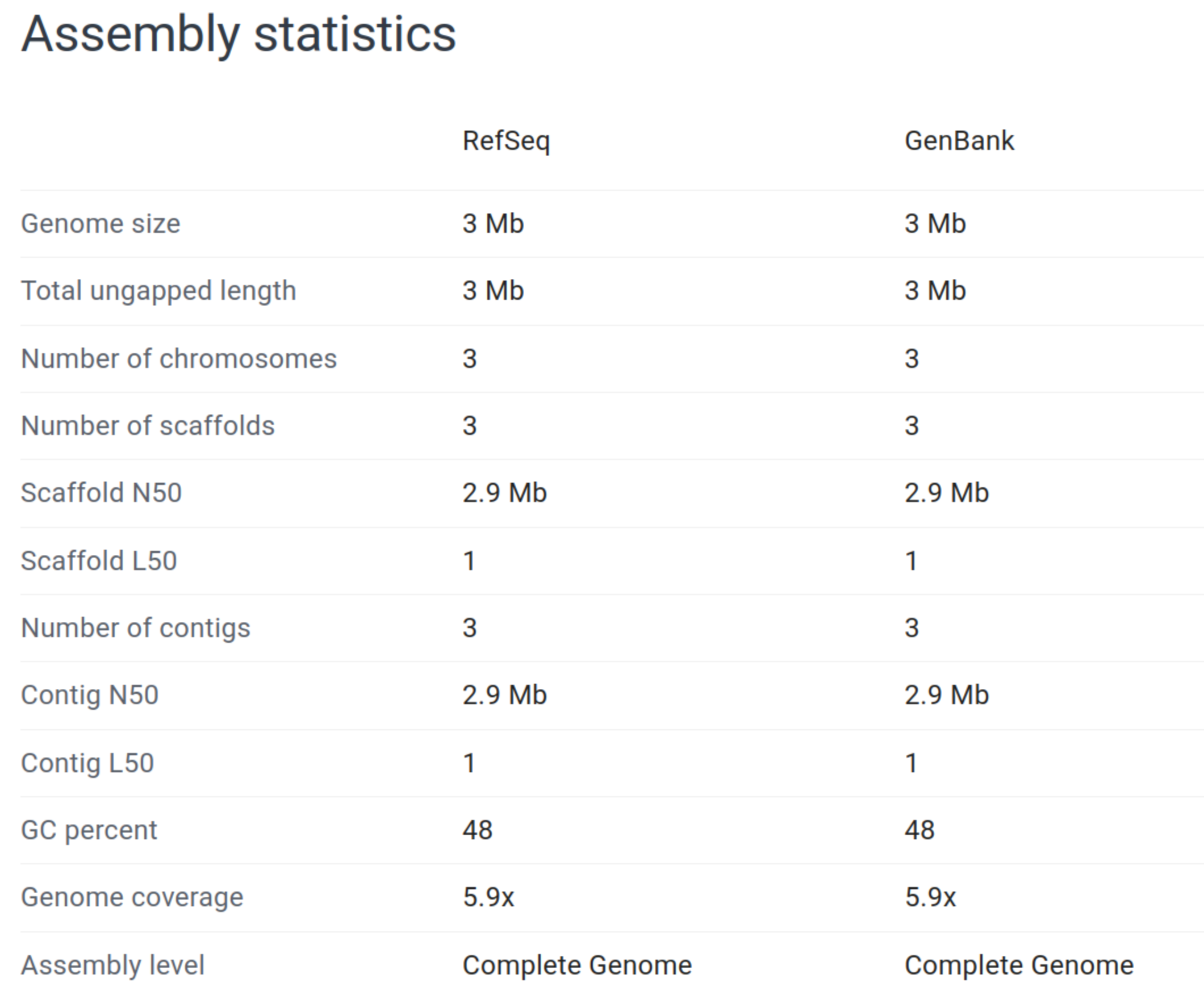
Galaxy13-fna.fasta FASTA DNA 449 3,116,159 227 6,940.2 805,683The assembly consists of 449 contigs, with a length of 3.1 Mb. Assembly length is close to that of other strains, including ASM82905v1
cd ~/work/PROJECTS/CAREMBOUCHE/
mkdir QUAST
conda activate quast-5.0.2 && HOME=/projet/tmp/ && quast.py Galaxy13-fna.fasta -o QUAST --min-contig 10 -r REF/GCF_000829055.1_ASM82905v1_genomic.fna && conda deactivatemkdir BUSCO
cd BUSCO
conda activate busco-5.3.2
wget https://busco-data.ezlab.org/v5/data/lineages/lactobacillales_odb10.2024-01-08.tar.gz
tar zxvf lactobacillales_odb10.2024-01-08.tar.gz
2024-06-19 11:42:26 INFO: ***** Start a BUSCO v5.3.2 analysis, current time: 06/19/2024 11:42:26 *****
2024-06-19 11:42:26 INFO: Configuring BUSCO with local environment
2024-06-19 11:42:26 INFO: Mode is genome
2024-06-19 11:42:26 INFO: Downloading information on latest versions of BUSCO data...
2024-06-19 11:42:26 INFO: Input file is /work_home/orue/PROJECTS/CAREMBOUCHE/Galaxy13-fna.fasta
2024-06-19 11:42:26 INFO: Downloading file 'https://busco-data.ezlab.org/v5/data/lineages/lactobacillales_odb10.2024-01-08.tar.gz'
2024-06-19 11:42:28 INFO: Decompressing file '/work_home/orue/PROJECTS/CAREMBOUCHE/BUSCO/busco_downloads/lineages/lactobacillales_odb10.tar.gz'
2024-06-19 11:42:31 INFO: Running BUSCO using lineage dataset lactobacillales_odb10 (prokaryota, 2024-01-08)
2024-06-19 11:42:31 INFO: ***** Run Prodigal on input to predict and extract genes *****
2024-06-19 11:42:31 INFO: Running Prodigal with genetic code 11 in single mode
2024-06-19 11:42:31 INFO: Running 1 job(s) on prodigal, starting at 06/19/2024 11:42:31
2024-06-19 11:42:37 INFO: [prodigal] 1 of 1 task(s) completed
2024-06-19 11:42:37 INFO: Genetic code 11 selected as optimal
2024-06-19 11:42:37 INFO: ***** Run HMMER on gene sequences *****
2024-06-19 11:42:37 INFO: Running 402 job(s) on hmmsearch, starting at 06/19/2024 11:42:37
2024-06-19 11:42:40 INFO: [hmmsearch] 41 of 402 task(s) completed
2024-06-19 11:42:40 INFO: [hmmsearch] 81 of 402 task(s) completed
2024-06-19 11:42:40 INFO: [hmmsearch] 121 of 402 task(s) completed
2024-06-19 11:42:41 INFO: [hmmsearch] 161 of 402 task(s) completed
2024-06-19 11:42:41 INFO: [hmmsearch] 202 of 402 task(s) completed
2024-06-19 11:42:41 INFO: [hmmsearch] 242 of 402 task(s) completed
2024-06-19 11:42:42 INFO: [hmmsearch] 282 of 402 task(s) completed
2024-06-19 11:42:42 INFO: [hmmsearch] 322 of 402 task(s) completed
2024-06-19 11:42:42 INFO: [hmmsearch] 362 of 402 task(s) completed
2024-06-19 11:42:43 INFO: [hmmsearch] 402 of 402 task(s) completed
2024-06-19 11:42:43 INFO: Results: C:99.3%[S:98.8%,D:0.5%],F:0.7%,M:0.0%,n:402
2024-06-19 11:42:46 INFO:
--------------------------------------------------
|Results from dataset lactobacillales_odb10 |
--------------------------------------------------
|C:99.3%[S:98.8%,D:0.5%],F:0.7%,M:0.0%,n:402 |
|399 Complete BUSCOs (C) |
|397 Complete and single-copy BUSCOs (S) |
|2 Complete and duplicated BUSCOs (D) |
|3 Fragmented BUSCOs (F) |
|0 Missing BUSCOs (M) |
|402 Total BUSCO groups searched |
--------------------------------------------------
2024-06-19 11:42:46 INFO: BUSCO analysis done. Total running time: 19 seconds
2024-06-19 11:42:46 INFO: Results written in /work_home/orue/PROJECTS/CAREMBOUCHE/BUSCO/busco_output
2024-06-19 11:42:46 INFO: For assistance with interpreting the results, please consult the userguide: https://busco.ezlab.org/busco_userguide.html
2024-06-19 11:42:46 INFO: Visit this page https://gitlab.com/ezlab/busco#how-to-cite-busco to see how to cite BUSCO
generate_plot.py -wd busco_output/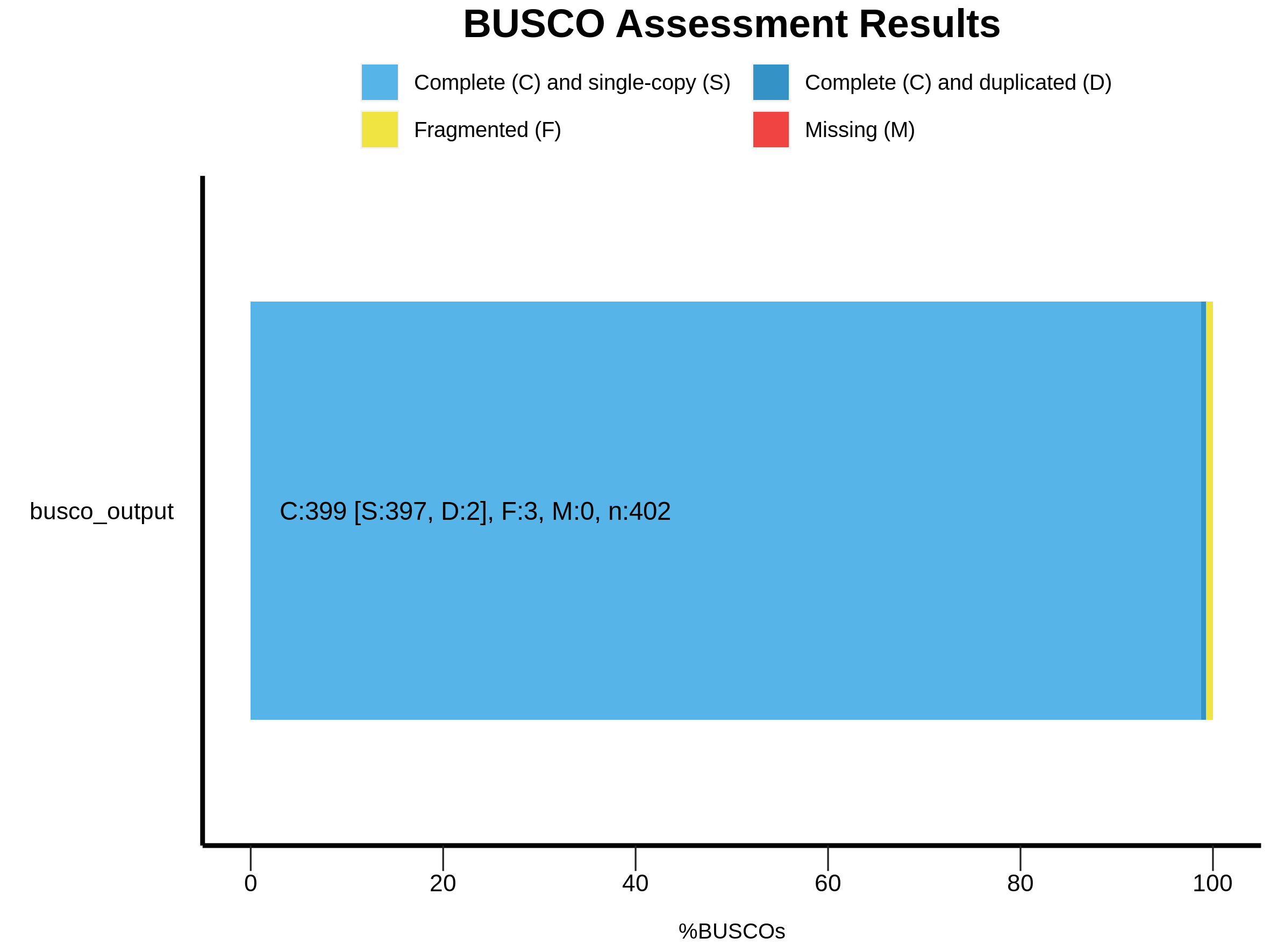
cd /home/orue/work/PROJECTS/CAREMBOUCHE/CHECKM
conda activate checkm2-0.1.3
checkm2 predict --threads 16 --input ../SPADES/BUSCO/busco_output/prodigal_output/predicted_genes/predicted.faa --output-directory . --genes
more quality_report.tsv
Name Completeness Contamination Completeness_Model_Used Additional_Notes
predicted 99.76 1.13 Neural Network (Specific Model) NonePaul sent me a sequence (https://www.ncbi.nlm.nih.gov/protein/AFJ15093.1) found in a publication where the authors are looking for putative Prt genes in the genome of L. casei by blasting this sequence.
This assembly is very fragmented (449 contigs). Less a genome is fragmented, better is theorically the gene annotation.
I suggested to Paul that I test other assemblers myself to try to improve the assembly.
I got the FASTQ files in the Galaxy of Gladys. They contain about 4 M reads (150 bp). I tested different assemblers.
cd /home/orue/work/PROJECTS/CAREMBOUCHE/UNICYCLER
conda activate unicycler-0.5.0
unicycler -1 ../FASTQ/data_R1.fastq.gz -2 ../FASTQ/data_R2.fastq.gz -o . --keep 0 -t 16
grep -c '>' /work_home/orue/PROJECTS/CAREMBOUCHE/UNICYCLER/assembly.fasta
38conda activate spades-3.15.3
spades.py -1 ../FASTQ/data_R1.fastq.gz -2 ../FASTQ/data_R2.fastq.gz -k 11,17,25,31,43,55,77,81 -t 16 -o . --isolate
conda deactivate| tool | contigs | total length | N50 | largest contig | CDS |
|---|---|---|---|---|---|
| Original (Spades single-end) | 449 | 3116159 | 234751 | 805683 | 2778 |
| Spades | 475 | 3124564 | 257129 | 848256 | 2777 |
| Megahit | 85 | 3026066 | 234879 | 805707 | 2764 |
| Unicycler | 38 | 3005286 | 403998 | 847367 | 2756 |
The assembly produced by Unicycler is less fragmented than others, for a similar assembly length and gene content. This assembly will be used as reference for now.
conda activate bakta-1.9.1
bakta --db /db/outils/bakta-1.9.1/db/ --output BAKTA/ assembly.fasta --threads 8mkdir /home/orue/work/PROJECTS/CAREMBOUCHE/UNICYCLER/DBCAN
cd /home/orue/work/PROJECTS/CAREMBOUCHE/UNICYCLER/
conda activate run_dbcan-3.0.2
#run_dbcan assembly.fasta prok --out_dir DBCAN --db_dir /db/outils/dbcan/ --tools all --hmm_cpu 16 --eCAMI_jobs 16 --cluster ../BAKTA/assembly.gff3
run_dbcan BAKTA/assembly.fna prok --out_dir DBCAN2 --db_dir /db/outils/dbcan/ --tools all --hmm_cpu 16 --eCAMI_jobs 16 --cluster BAKTA/assembly.gff3
egrep -v -e "^[ATCGN]" -e "^##FASTA" -e "^>contig" BAKTA/assembly.gff3 > BAKTA/assembly_without_seqs.gff3
python ~/save/SCRIPTS/add_dbcan_to_gff3.py --gff BAKTA/assembly.gff3 --overview DBCAN2/overview.txt --gff-prodigal DBCAN2/prodigal.gff --output dbcan2.gff3 --cgc-file DBCAN2/cgc_standard.outA lactocepin sequence, see ref was then blasted against the genes with blastp [9]. This sequence was previously used to find putative Prt genes in L. casei genomes.
cd /home/orue/work/PROJECTS/CAREMBOUCHE/UNICYCLER
mkdir BLAST
conda activate blast-2.15.0
makeblastdb -in BAKTA/assembly.faa -dbtype prot
cd BLAST
blastp -query ../../paul_sequence_putative_prt_lcassei.prot.fasta -db ../BAKTA/assembly.faa -outfmt "6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore stitle" -out alignment.outHere is the location of the best hit: contig_2 9923 11812
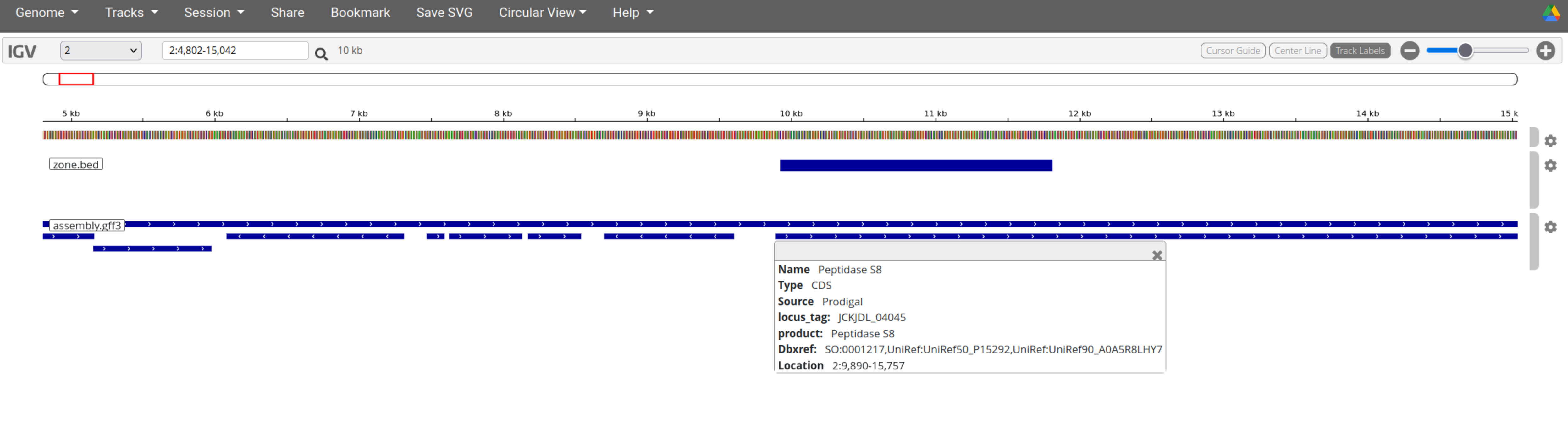
The sequence is located in a CDS annotated Peptidase S8.
cd /home/orue/work/PROJECTS/CAREMBOUCHE/UNICYCLER
mkdir EGGNOG
cd EGGNOG
conda activate eggnog-mapper-2.1.5
emapper.py -i ../BAKTA/assembly.faa --output eggnog -m diamond -d bact --cpu 16 --data_dir /db/outils/eggnog-mapper-2.1.5/ --output_dir . --decorate_gff ../dbcan2.gff3
conda deactivatePaul pointed me the MEROPS [11] database for peptidases annotation. The MEROPS database is a resource for information on peptidases (sometimes also termed proteases, proteinases and proteolytic enzymes).
Its use is complicated because it required to build a MySQL instance. Nevertheless, MEROPS gives the possibility to download some flat files:
The file “pepunit.lib” is a non-redundant library of protein sequences in FastA format the peptidase units and inhibitor units of all the peptidases and peptidase inhibitors that are included in MEROPS. The library can be searched by use of FastA without further modification, but must be converted and indexed for BLAST searches. The restriction of the library to peptidase and inhibitor units should decrease the risk of false positive matches to other domains.
cd /home/orue/work/PROJECTS/CAREMBOUCHE
wget https://ftp.ebi.ac.uk/pub/databases/merops/current_release/pepunit.lib # Download file
grep -c '>' pepunit.lib
1227939The file contains 1,227,939 proteic sequences. Information is found in the sequence header.
head -n 2 pepunit.lib
>MER0000001 - chymotrypsin B (Homo sapiens) [S01.152]#S01A#{peptidase unit: 34-263}~source CTRB_HUMAN~
IVNGEDAVPGSWPWQVSLQDKTGFHFCGGSLISEDWVVTAAHCGVRTSDVVVAGEFDQGSI used seqkit [12] to find sequences with some patterns: PrtP, PrtS, Lactobacillus, paracasei and casei.
conda activate seqkit-2.0.0
seqkit grep -r -p Lactobacillus -p paracasei -p casei -p PrtP -p Prtp -p prtp -n pepunit.lib > pepunit_lactocasei_and_prtp.lib
grep -c '>' pepunit_lactocasei_and_prtp.lib
8058and then used fasta36 [13] to align proteic sequences predicted with Bakta against the MEROPS reduced databank.
mkdir MEROPS
conda activate fasta-36.3.8e
fasta36 BAKTA/assembly.faa ../pepunit_lactocasei_and_prtp.lib -m 8 -E 0.05 -T 16 > MEROPS/pepunit_lactocasei_and_prtp_fasta36.out
wc -l pepunit_lactocasei_and_prts_fasta36.out
13072The output file contains information about alignments:
grep ">" pepunit_lactocasei_and_prtp.lib | sed "s/\ -\ /;/g" | sed "s/>//g" > pepunit_lactocasei_and_prtp.lib.infosI join now the information about subjects
alignments <- vroom::vroom("data/pepunit_lactocasei_and_prtp_fasta36.out", col_names=F)
colnames(alignments) <- c("QueryID","SubjectID","Percidentity","AlignmentLength","Mismatches","GapOpenings","QueryStart","QueryEnd","SubjectStart","SubjectEnd","E-value","BitScore")
# on ajoute les infos trouvées dans les headers
infos <- vroom::vroom("data/pepunit_lactocasei_and_prtp.lib.infos", col_names=F, delim = ";")
colnames(infos) <- c("ID","Details")
mytable <- left_join(alignments,infos,by=join_by(SubjectID==ID))
library(reactable)
reactable::reactable(mytable, columns = list(
Details = colDef(width = 200)
))You can look in the GFF3 file where hits fall by searching for the query ID. For example the annotation of JCKJDL_02300 is ID=JCKJDL_02300;Name=putative metalloendopeptidase;locus_tag=JCKJDL_02300;product=putative metalloendopeptidase;Dbxref=COG:COG3590,COG:O,RefSeq:WP_049169840.1,SO:0001217,UniParc:UPI0006674A97,UniRef:UniRef100_UPI0006674A97,UniRef:UniRef50_K0N8Q0,UniRef:UniRef90_A0A179YD63;gene=pepO
library(rtracklayer)
gff <- readGFF("html/eggnog.emapper.gff")A work by Migale Bioinformatics Facility
Université Paris-Saclay, INRAE, MaIAGE, 78350, Jouy-en-Josas, France
Université Paris-Saclay, INRAE, BioinfOmics, MIGALE bioinformatics facility, 78350, Jouy-en-Josas, France
