
cd /home/orue/work/16SIONTORRENT/
mkdir RAW_DATA
cp /home/mlavie/work/Limoges_IonT/RAW_DATA/Puce1/IonXpress_033_J0M1_rawlib.basecaller.fq RAW_DATA/
for i in *.fq ; do id=$(echo $i |cut -d '_' -f 2-3) ; mv $i ${id}.fastq ; done
pigz *.fastq
tar zcvf 16SIonTorrent.tar.gz *.fastq.gz16SIonTorrent
Bioinformatic analysis of IonTorrent data
closed
collaboration
Note
This document is a report of the analyses performed. You will find all the code used to analyze these data. The version of the tools (maybe in code chunks) and their references are indicated, for questions of reproducibility.
Aim of the project
The aim of these analyses is to obtain a BIOM file from 16S Ion Torrent data.
Data management
Important
All data is managed by the migale facility for the duration of the project. Once the project is over, the Migale facility does not keep your data. We will provide you with the raw data and associated metadata that will be deposited on public repositories before the results are used. We can guide you in the submission process. We will then decide which files to keep, knowing that this report will also be provided to you and that the analyses can be replayed if needed.
Sequencing data
Data were available in the Mathias working directory. We copied them, renamed them, compressed them and stored them in an archive.
# seqkit
cd /home/orue/work/16SIONTORRENT/
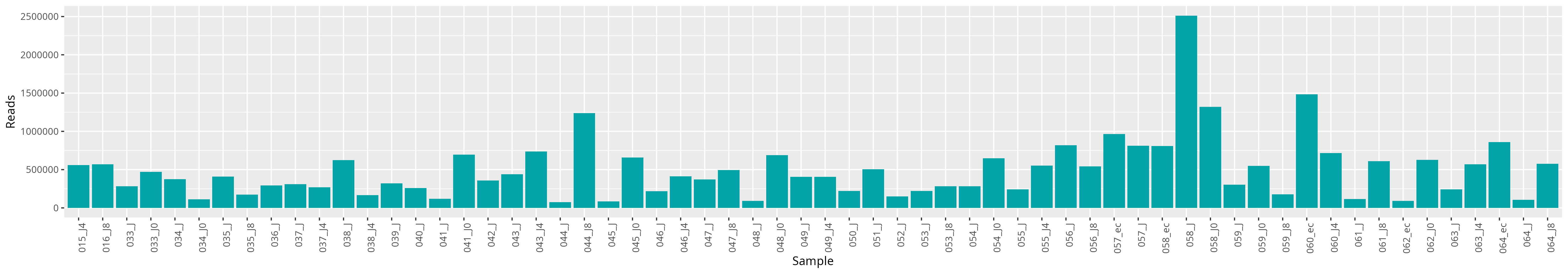
qsub -cwd -V -N seqkit -q maiage.q -pe thread 4 -R y -b y "conda activate seqkit-2.0.0 && seqkit stats /home/orue/work/16SIONTORRENT/RAW_DATA/*.fastq.gz -j 4 > raw_data.infos && conda deactivate"We can plot and display the number of reads to see if enough reads are present and if samples are homegeneous.
Quality control
cd /home/orue/work/16SIONTORRENT/
mkdir FASTQC LOGS
for i in /home/orue/work/16SIONTORRENT/RAW_DATA/*.fastq.gz ; do echo "conda activate fastqc-0.11.9 && fastqc $i -o FASTQC && conda deactivate" >> fastqc.sh ; done
qarray -cwd -V -N fastqc -o LOGS -e LOGS fastqc.sh
qsub -cwd -V -N multiqc -o LOGS -e LOGS -b y "conda activate multiqc-1.11 && multiqc FASTQC -o MULTIQC && conda deactivate"
Note
Quality control shows heterogeneous metrics between samples. Some samples are very poorly sequenced (controls but also samples of interest). The sequencing quality of some samples is also poor after 150 base pairs. There are still some N’s in a few reads, we also notice the presence of Illumina adapters, indicating very small fragments. All of these poor quality reads will be discarded with bioinformatics.
Bioinformatics
Raw reads needed to be processed to build operational taxonomic units (OTUs). The FROGS [4] worfklow was used following authors guidelines.
tar zcvf 16SIonTorrent.tar.gz *.fastq.gz
preprocess.py illumina --min-amplicon-size 50 --max-amplicon-size 10000 --without-primers --already-contiged --input-archive 16SIonTorrent.tar.gz --nb-cpus 16Preprocess
The options --already-contiged and without-primers are used to deal with IonTorrent data.
preprocess.py illumina --min-amplicon-size 50 --max-amplicon-size 10000 --without-primers --already-contiged --input-archive RAW_DATA/16SIonTorrent.tar.gz --nb-cpus 16Clustering
clustering.py --input-fasta preprocess.fasta --input-count preprocess_counts.tsv --nb-cpus 2410,531,600 OTUs were built with 50,166,330 sequences.
Remove chimera
remove_chimera.py --input-fasta clustering_seeds.fasta --input-biom clustering_abundance.biom --nb-cpus 24 --summary remove_chimera.html
otu_filters.py --input-fasta remove_chimera.fasta --input-biom remove_chimera_abundance.biom --output-fasta remove_chimera.fastaFew chimera are detected.
OTU filters
otu_filters.py --input-fasta clustering_seeds.fasta --input-biom clustering_abundance.biom --nb-cpus 16 --log-file filters.log --output-biom filters.biom --summary filters.html --excluded filters_excluded.tsv --contaminant /db/outils/FROGS/contaminants/phi.fa --min-sample-presence 1 --min-abundance 0.00005 --output-fasta filters.fasta
Warning
A lot of OTUs are removed but reducing the threshold to 1,000 for example would not change a lot.
Taxonomic affiliation
affiliation_OTU.py --input-biom filters.biom --input-fasta filters.fasta --reference /db/outils/FROGS/assignation/silva_138.1_16S_pintail100/silva_138.1_16S_pintail100.fasta --nb-cpus 16
Warning
A lot of OTUs are not affiliated to Species level. There is not a lot of multi-affiliations but the affiliations are not precise in the databank (unknown species…)
Tree
tree.py --input-sequences filters.fasta --biom-file affiliation_abundance.biom --out-tree tree.nwkDownloads
References
1. Shen W, Le S, Li Y, Hu F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/q file manipulation. PloS one. 2016;11:e0163962.
2. Andrews S. FastQC a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
3. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–8.
4. Escudié F, Auer L, Bernard M, Mariadassou M, Cauquil L, Vidal K, et al. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics. 2018;34:1287–94. doi:10.1093/bioinformatics/btx791.
Reuse
This document will not be accessible without prior agreement of the partners
