
cd /home/orue/work/PMPBACT
mkdir RAW_DATA
# Rename files
for i in INSERM_C.Cocheray/*R1_001.fastq.gz ; do id=$(echo $(basename $i |cut -d '_' -f 1)) ; cp $i RAW_DATA/${id}_R1.fastq.gz ; done
for i in INSERM_C.Cocheray/*R2_001.fastq.gz ; do id=$(echo $(basename $i |cut -d '_' -f 1)) ; cp $i RAW_DATA/${id}_R2.fastq.gz ; done
cd RAW_DATA/
tar zcvf PMPBACT.tar.gz *.fastq.gz
# Save
scp PMPBACT.tar.gz abaca:/backup/partage/migale/PMPBACT/RAW_DATA/PMPBACT
Bacterial composition of rare peritoneal tumors in humans and mouses.
closed
collaboration
Note
This document is a report of the analyses performed. You will find all the code used to analyze these data. The version of the tools (maybe in code chunks) and their references are indicated, for questions of reproducibility.
Aim of the project
The aims of these analyses are to build operational taxonomic units (OTUs) from raw reads and to affiliate OTUs sequences to obtain the taxonomic composition of the samples. Reads were provided by the @BRIDGE from an Illumina Miseq instrument (2x251 bp). The targeted amplicon is the V3-V4 region of the 16S rRNA.
Data management
Important
All data is managed by the migale facility for the duration of the project. Once the project is over, the Migale facility does not keep your data. We will provide you with the raw data and associated metadata that will be deposited on public repositories before the results are used. We can guide you in the submission process. We will then decide which files to keep, knowing that this report will also be provided to you and that the analyses can be replayed if needed.
Sequencing data
Data were downloaded from Filesender, then deposited on the Front server and a copy was sent to the abaca.
seqkit
# seqkit
qsub -cwd -V -N seqkit -q maiage.q -pe thread 4 -R y -b y "conda activate seqkit-2.0.0 && seqkit stats *.fastq.gz -j 4 > raw_data.infos && conda deactivate"From this result, we plot the number of reads to see if enough reads are present and if samples are homegeneous.
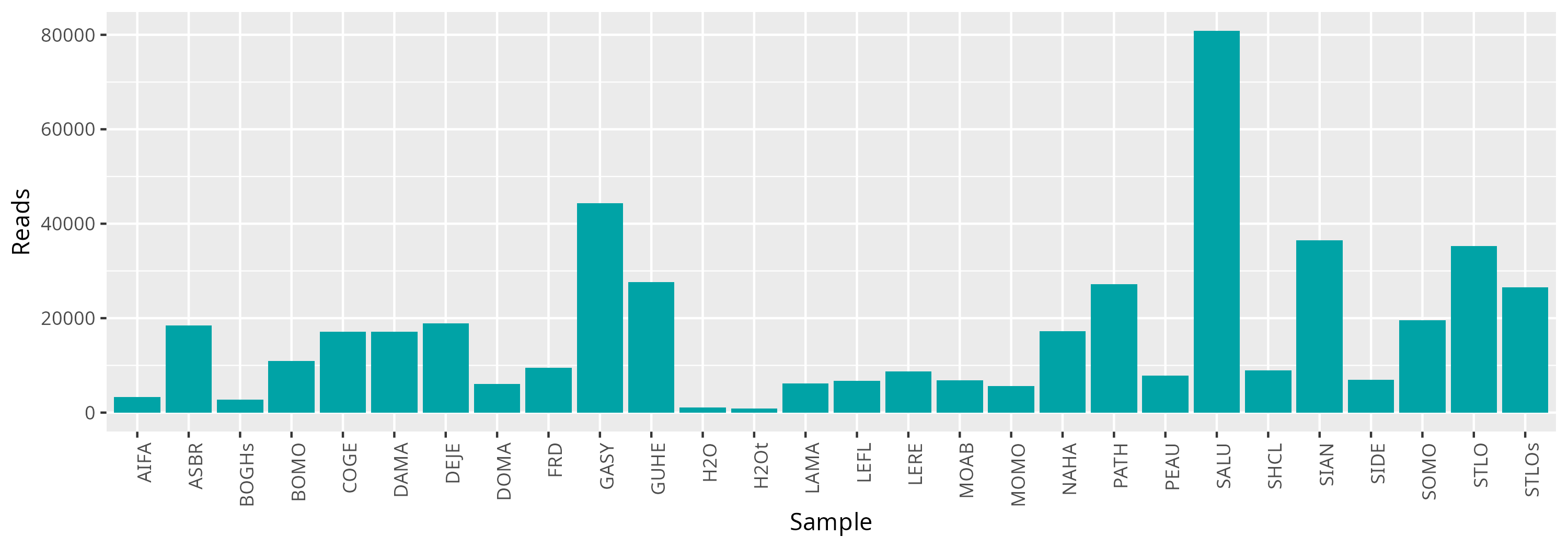
The information for all samples are indicated in the following table.
Warning
Some samples have very few reads. The sequencing effort may not be sufficient to detect a real signal
Quality control
The quality was assessed with FastQC
mkdir FASTQC LOGS
for i in RAW_DATA/*.fastq.gz ; do echo "conda activate fastqc-0.11.9 && fastqc $i -o FASTQC && conda deactivate" >> fastqc.sh ; done
qarray -cwd -V -N fastqc -o LOGS -e LOGS fastqc.sh
qsub -cwd -V -N multiqc -o LOGS -e LOGS -b y "conda activate multiqc-1.11 && multiqc FASTQC -o MULTIQC && conda deactivate"Here is the MultiQC report. We can see that all reads have the same size (251). The sequence quality histograms are expected for Miseq sequencing. The adapter conent show high proportion for some samples (H20 and LEFL samples). It means the fragments were too small. Those reads will be discarded later.
Note
The quality of the sequencing, assessed by standard NGS tools, is good enough to process the data.
Bioinformatics
FROGS
Preprocess
The preprocess tool aims at cleaning and dereplicating sequences. Reads are merged with pear
mkdir FROGS
cd FROGS
mkdir LOGS
qsub -cwd -V -N preprocess -o LOGS -e LOGS -pe thread 8 -R y -b y "conda activate frogs-4.0.1 && preprocess.py illumina --input-archive /work_home/orue/PMPBACT/RAW_DATA/PMPBACT.tar.gz --min-amplicon-size 200 --max-amplicon-size 490 --merge-software pear --five-prim-primer ACGGRAGGCAGCAG --three-prim-primer AGGATTAGATACCCTGGTA --R1-size 250 --R2-size 250 --nb-cpus 8 --output-dereplicated preprocess.fasta --output-count preprocess.tsv --summary preprocess.html --log-file preprocess.log && conda deactivate"The preprocess report shows that ~93% of the reads overlap, have the expected size and do not have ambiguous bases. 392,585 sequences are remaining (from 470,880).
Clustering
The clustering tool uses swarm
qsub -cwd -V -N clustering -o LOGS -e LOGS -pe thread 8 -R y -b y "conda activate frogs-4.0.1 && clustering.py --input-fasta preprocess.fasta --input-count preprocess.tsv --distance 1 --fastidious --nb-cpus 8 --log-file clustering.log --output-biom clustering.biom --output-fasta clustering.fasta --output-compo clustering_otu_compositions.tsv && conda deactivate"
qsub -cwd -V -N clusters_stats -o LOGS -e LOGS -b y "conda activate frogs-4.0.1 && clusters_stat.py --input-biom clustering.biom --output-file clusters_stats.html --log-file clusters_stats.log && conda deactivate"The clustering statistics report shows that the 392,585 sequences are grouped into 28,060 OTUs. 96% of the OTUs are composed of only 1 sequence and will be removed later. The biggest OTU is composed of 65,717 sequences.
Chimera removal
Chimeras are sequences formed from two or more biological sequences joined together. The majority of these anomalous sequences are formed from an incomplete extension during a PCR cycle. During subsequent cycles, a partially extended strand can bind to a template derived from a different but similar sequence. This phenomena is particularly common in amplicon sequencing where closely related sequences are amplified. The chimera detection is performed with vsearch
qsub -cwd -V -N chimera -o LOGS -e LOGS -pe thread 8 -R y -b y "conda activate frogs-4.0.1 && remove_chimera.py --input-fasta clustering.fasta --input-biom clustering.biom --non-chimera remove_chimera.fasta --nb-cpus 8 --log-file remove_chimera.log --out-abundance remove_chimera.biom --summary remove_chimera.html && conda deactivate"The specific report shows that 1.1% of the sequences are detected as chimeric sequences. It corresponds to 4% of the OTUs.
Filters on abundance
Here, we applied only a filter on abundance (not on prevalence). We applied the classical threshold of 0.005% of overall abundance (corresponding to 20 sequences). We checked also if phiX sequences still remain in sequences.
qsub -cwd -V -N filters -o LOGS -e LOGS -pe thread 8 -R y -b y "conda activate frogs-4.0.1 && otu_filters.py --input-fasta remove_chimera.fasta --input-biom remove_chimera.biom --output-fasta filters.fasta --nb-cpus 8 --log-file filters.log --output-biom filters.biom --summary filters.html --excluded filters_excluded.tsv --contaminant /db/outils/FROGS/contaminants/phi.fa --min-sample-presence 1 --min-abundance 0.00005 && conda deactivate"The report shows that 186 OTUs are remaining (99.3% of OTUs are removed), corresponding to 360,732 sequences (92.9%). No phiX contamination is present. Everything is normal.
Taxonomic assignation
We now performed a taxonomic assignation of OTUs with blast
qsub -cwd -V -N affiliation -o LOGS -e LOGS -pe thread 8 -R y -b y "conda activate frogs-4.0.1 && affiliation_OTU.py --input-fasta filters.fasta --input-biom filters.biom --nb-cpus 8 --output-biom affiliation.biom --summary affiliation.html --reference /db/outils/FROGS/assignation/silva_138.1_16S_pintail100/silva_138.1_16S_pintail100.fasta && conda deactivate"
qsub -cwd -V -N affiliations_stats -o LOGS -e LOGS -b y "conda activate frogs-4.0.1 && affiliations_stat.py --input-biom affiliation.biom --output-file affiliations_stats.html --log-file affiliations_stats.log --multiple-tag blast_affiliations --tax-consensus-tag blast_taxonomy --identity-tag perc_identity --coverage-tag perc_query_coverage && conda deactivate"This report shows that 74% of the OTUs are affiliated (96% of sequences). 62% of the affiliated OTUs are multi-affiliated at Species level (the sequence may be not discriminating enough to differentiate between several species of the same genus). The alignment distribution tab in this report shows that blast hits demonstrate all a high identity and coverage percents, except 2 with 6676 sequences. The affiliation of these OTUs have to be checked.
Note
Species can not be assigned for a lot of OTUs. It is expected for 16S sequences because 16S rRNA sequence is often the same between Species.
Finally, we created a TSV file from the BIOM file.
qsub -cwd -V -N biom_to_tsv -o LOGS -e LOGS -b y "conda activate frogs-4.0.1 && biom_to_tsv.py --input-biom affiliation.biom --input-fasta filters.fasta --output-tsv affiliation.tsv --output-multi-affi multi_aff.tsv --log-file biom_to_tsv.log && conda deactivate"Informations are displayed in the following table:
Some sequences were not affiliated against Silva. We checked manually from the NCBI blast website their taxonomic affiliation. An example is available in the following screenshot. OTUs without affiliation are human or mouse sequences.
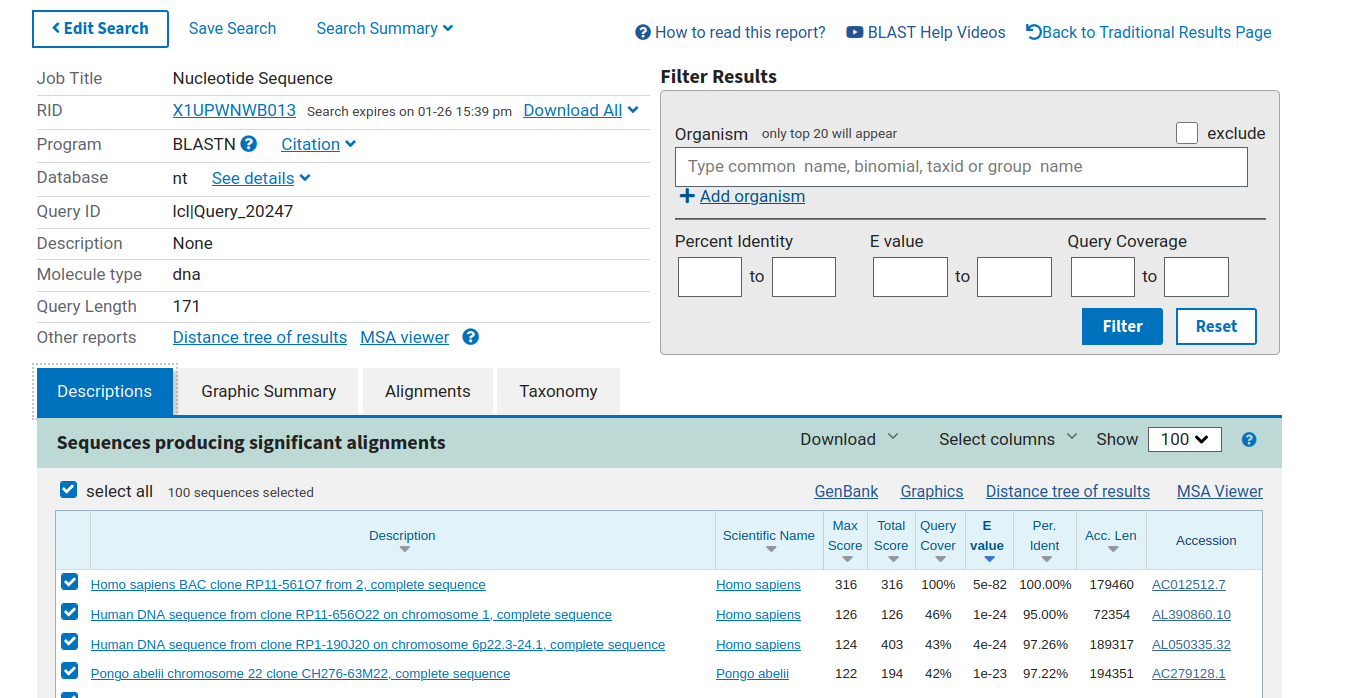
We removed them with the otu_filters tool (we chose the human genome as contaminant) and checked the result.
qsub -cwd -V -N filters -o LOGS -e LOGS -pe thread 8 -R y -b y "conda activate frogs-4.0.1 && otu_filters.py --contaminant hs1.fa --input-biom affiliation.biom --input-fasta filters.fasta --output-biom affiliation2.biom --output-fasta filters2.fasta --summary filters2.html --log-file filters2.log --excluded filters2_excluded.tsv && conda deactivate"
qsub -cwd -V -N affiliations_stats2 -o LOGS -e LOGS -b y "conda activate frogs-4.0.1 && affiliations_stat.py --input-biom affiliation2.biom --output-file affiliations_stats2.html --log-file affiliations_stats2.log --multiple-tag blast_affiliations --tax-consensus-tag blast_taxonomy --identity-tag perc_identity --coverage-tag perc_query_coverage && conda deactivate"The updated report shows that some OTUs without affiliation are still present. They (Cluster_117, Cluster_115, Cluster_202, Cluster_214, Cluster_67, CLuster_111) are human and mouse sequences. We have removed them later.
Here are the multiple affiliations found for each multi-affiliated OTU:
Tip
AffiliationExplorer can be used to easily deal with multi-affiliations. The aim is to check if some multi-affiliations could be corrected.
Phylogenetic tree
We built a phylogenetic tree by performing a multiple alignment of OTUs with Mafft
qsub -cwd -V -N tree -o LOGS -e LOGS -pe thread 8 -R y -b y "conda activate frogs-4.0.1 &&tree.py --input-sequences filters2.fasta --biom-file affiliation2.biom --out-tree tree.nwk && conda deactivate"Biostatistics
We used phyloseq
Here are the informations about the samples:
| sample | Host | Column | Grade | Sex | Age | PCI |
|---|---|---|---|---|---|---|
| STLOs | souris | 1 | HG | NA | NA | NA |
| BOGHs | souris | 1 | HG | NA | NA | NA |
| DOMA | humain | 1 | HG | F | 69 | NA |
| STLO | humain | 1 | HG | M | NA | NA |
| PATH | humain | 1 | HG | M | 47 | NA |
| GUHE | humain | 1 | HG | M | 64 | 39 |
| GASY | humain | 1 | HG | F | 52 | NA |
| SOMO | humain | 1 | HG | F | 72 | NA |
| MOAB | humain | 2 | BG | M | 43 | 30 |
| SALU | humain | 2 | BG | F | 58 | NA |
| SIAN | humain | 2 | BG | M | NA | NA |
| DEJE | humain | 2 | BG | M | 50 | NA |
| COGE | humain | 2 | BG | M | 62 | NA |
| LAMA | humain | 2 | BG | F | 83 | NA |
| PEAU | humain | 2 | BG | F | 43 | NA |
| LERE | humain | 2 | BG | M | 73 | 39 |
| ASBR | humain | 3 | BG | M | 57 | NA |
| LEFL | humain | 3 | HG | F | 43 | NA |
| DAMA | humain | 3 | BG | F | 68 | NA |
| SIDE | humain | 3 | BG | F | 55 | NA |
| BOMO | humain | 3 | BG | F | NA | NA |
| SHCL | humain | 3 | BG | F | 56 | NA |
| MOMO | humain | 3 | NA | M | 72 | NA |
| AIFA | humain | 3 | BG | F | 62 | NA |
| NAHA | humain | 4 | BG | M | 46 | NA |
| FRD | humain | 4 | BG | F | NA | NA |
| H2O | control | 12 | NA | NA | NA | NA |
| H2Ot | control | 12 | NA | NA | NA | NA |
We removed OTUs without affiliations.
my_bad_otus <- c("Cluster_117", "Cluster_115", "Cluster_202", "Cluster_214", "Cluster_67", "Cluster_111")
allTaxa = taxa_names(physeq)
allTaxa_final <- allTaxa[!(allTaxa %in% my_bad_otus)]
physeq <- prune_taxa(allTaxa_final, physeq)6 OTUs have been removed.
Finally, we checked the amount of reads lost before and after all our analyses.
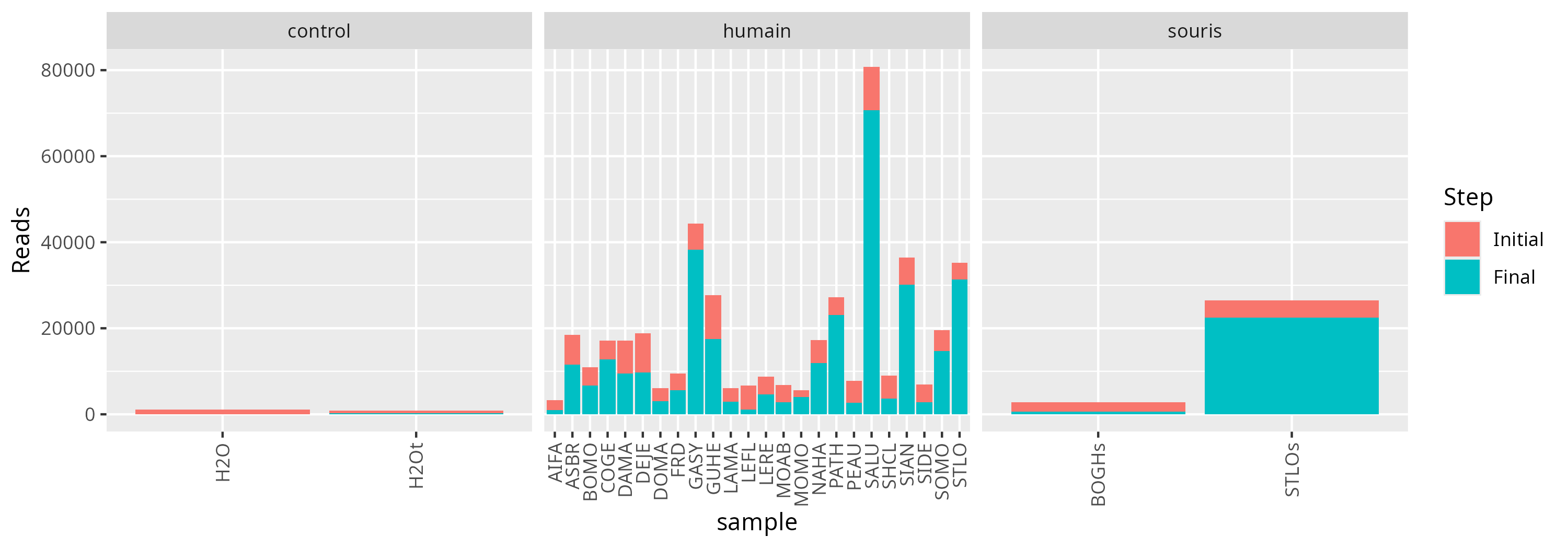
Note
Globally, 72.11% of reads is present in our count table.
Control samples
Let look at the composition of the control samples. The composition is different between them but represent very few reads. We can remove them.
physeq_controls <- subset_samples(physeq, Host=="control")
p <- plot_composition(physeq = physeq_controls, taxaRank1 = "Kingdom", taxaSet1 = "Bacteria", taxaRank2 = "Phylum", numberOfTaxa = 10L, x = "Sample")
physeq <- subset_samples(physeq, Host!="control")This is what our phyloseq object is made of:
physeqphyloseq-class experiment-level object
otu_table() OTU Table: [ 138 taxa and 26 samples ]
sample_data() Sample Data: [ 26 samples by 8 sample variables ]
tax_table() Taxonomy Table: [ 138 taxa by 7 taxonomic ranks ]
phy_tree() Phylogenetic Tree: [ 138 tips and 137 internal nodes ]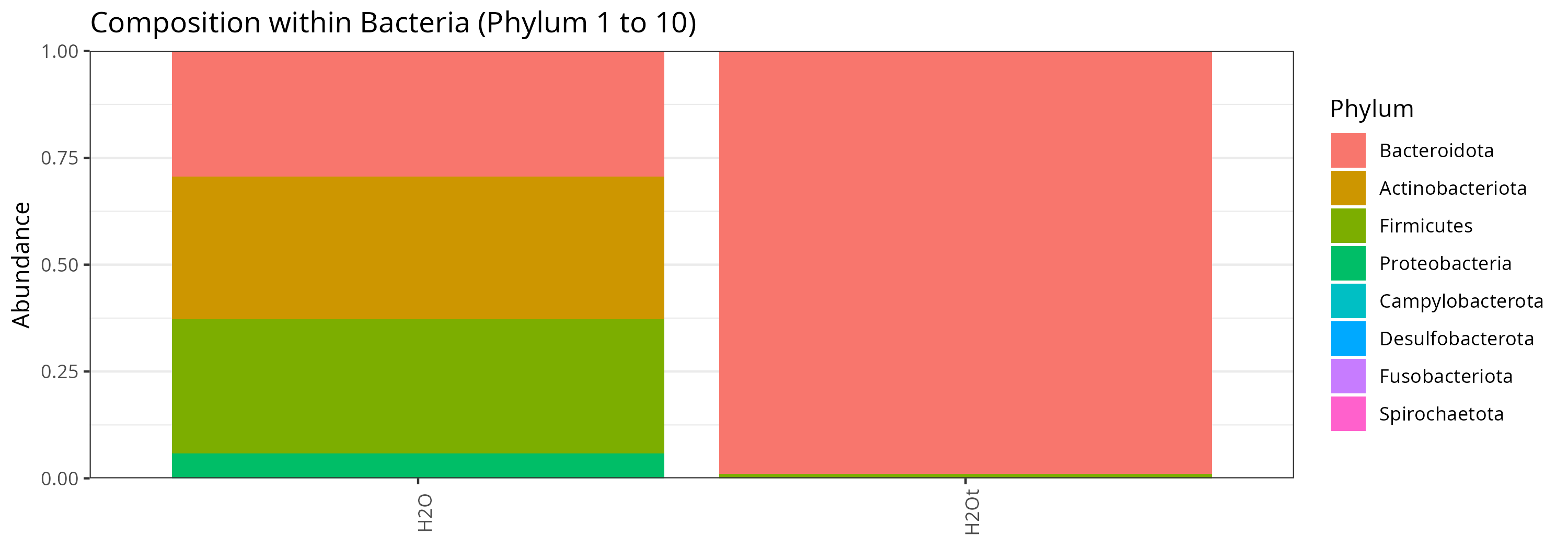
The final phyloseq object is avaialble here
Rarefaction curves
The depth is not homogenous between samples.
sample_sums(physeq) %>% sort()BOGHs AIFA LEFL PEAU SIDE MOAB LAMA DOMA SHCL MOMO LERE FRD BOMO
621 1038 1145 2644 2744 2799 2867 3094 3633 3961 4679 5549 6672
DAMA DEJE ASBR NAHA COGE SOMO GUHE STLOs PATH SIAN STLO GASY SALU
9506 9771 11565 11904 12703 14684 17469 22515 23033 30080 31398 38229 70692 and the rarefaction curves demonstrate it.
We test whether the depths used in this study are sufficient to capture the microbial diversity. The rarefaction do not reach a flat plateau for the majority of samples.
p <- ggrare(physeq, step = 500, se = FALSE, color = "Host", plot = FALSE, verbose = FALSE)
p <- p + geom_vline(xintercept = c(min(sample_sums(physeq)), 5000),
color = "grey50", linetype = "dashed") +
facet_grid(~ Host)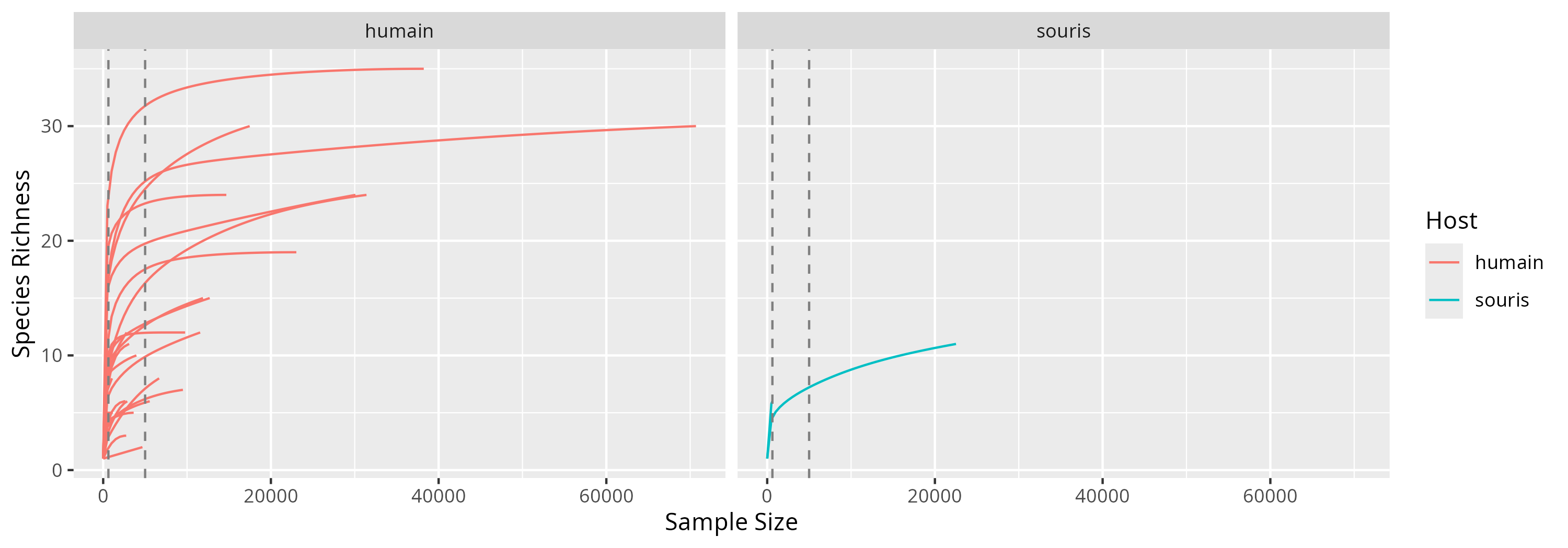
The shallowest samples clearly have too few reads to reflect the whole composition.
Alpha diversity
The observed richness is between a few OTUs and about 30.
p <- plot_richness(physeq = physeq, x = "samples", color = "Host", shape = NULL, title = "Alpha diversity graphics", measures = c("Observed"))
p <- p + geom_boxplot() + geom_point() 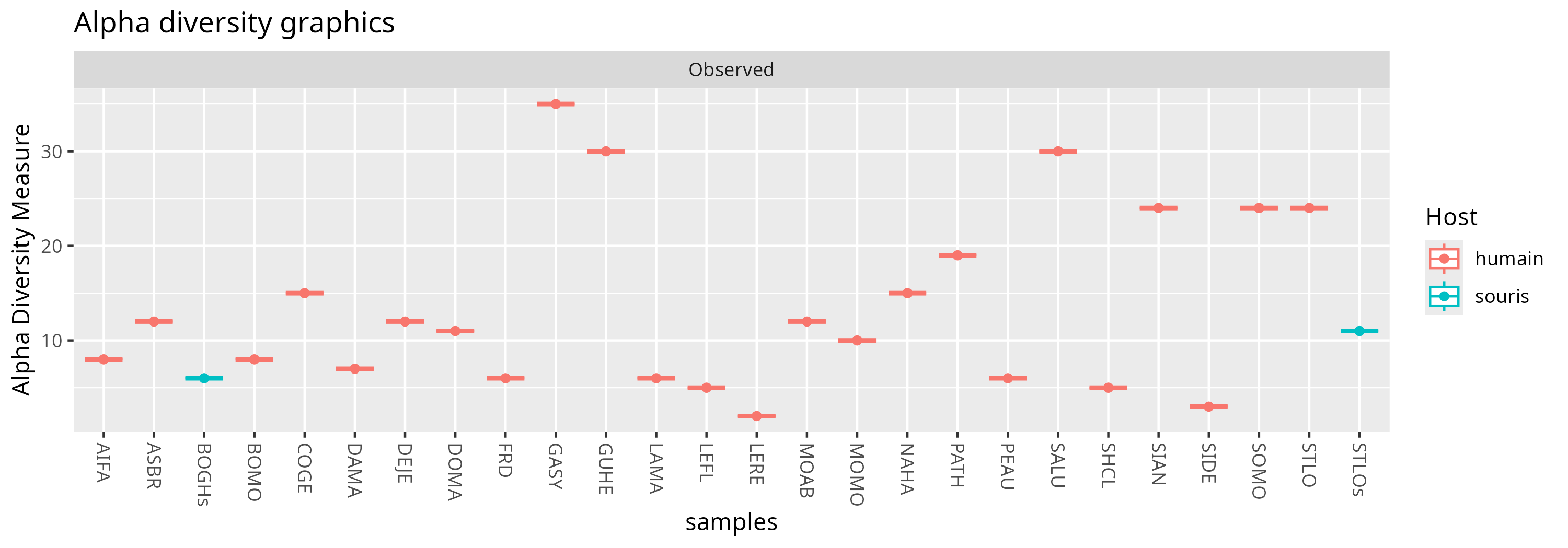
Compositions
We can look at the composition at the Phylum level
p <- plot_composition(physeq = physeq, taxaRank1 = "Kingdom", taxaSet1 = "Bacteria", taxaRank2 = "Phylum", numberOfTaxa = 10L, x = "Sample", title = "")
p <- p + facet_grid(". ~ Host", scales = "free_x", space = "free")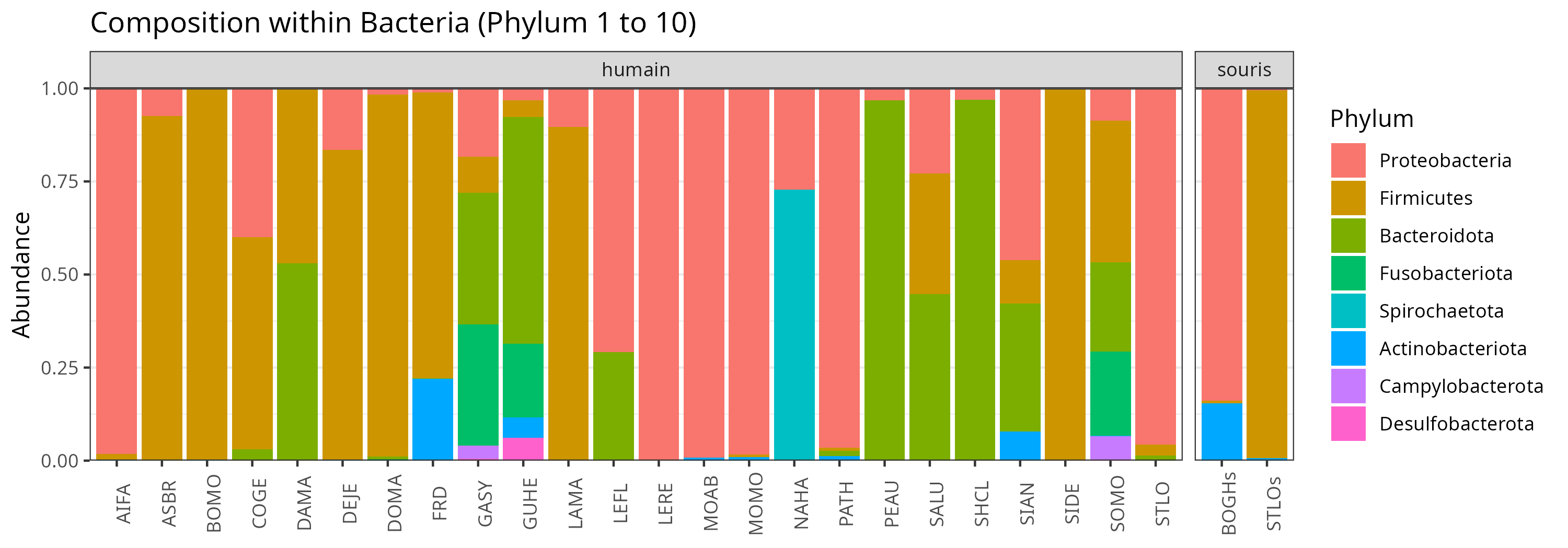
The samples composition is very different according to samples, even inside Human/Mouse samples.
Tip
To go further, Easy16S can be used with the rds file available in the Downloads section to explore in depth the results.
Downloads
References
References
1. Shen W, Le S, Li Y, Hu F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/q file manipulation. PloS one. 2016;11:e0163962.
2. Andrews S. FastQC a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
3. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–8.
4. Escudié F, Auer L, Bernard M, Mariadassou M, Cauquil L, Vidal K, et al. FROGS: Find, Rapidly, OTUs with Galaxy Solution. Bioinformatics. 2018;34:1287–94. doi:10.1093/bioinformatics/btx791.
5. Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: A fast and accurate illumina paired-end reAd mergeR. Bioinformatics. 2013;30:614–20.
6. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet journal. 2011;17:10–2.
7. Mahé F, Rognes T, Quince C, Vargas C de, Dunthorn M. Swarm v2: Highly-scalable and high-resolution amplicon clustering. PeerJ. 2015;3:e1420.
8. Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: A versatile open source tool for metagenomics. PeerJ. 2016;4:e2584.
9. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of molecular biology. 1990;215:403–10.
10. Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic acids research. 2012;41:D590–6.
11. Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular biology and evolution. 2013;30:772–80.
12. Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS one. 2010;5:e9490.
13. Schliep KP. Phangorn: Phylogenetic analysis in r. Bioinformatics. 2011;27:592–3.
14. McMurdie PJ, Holmes S. Phyloseq: An r package for reproducible interactive analysis and graphics of microbiome census data. PloS one. 2013;8:e61217.
