#Use packages and functionslibrary(tidyverse)library(data.table)library(SummarizedExperiment) # manipulation RNASeq assayslibrary(rtracklayer) ## for annotation#library("DEFormats")library(edgeR)library(kableExtra)library(dplyr)library(purrr)library(stringr)library(ggplot2)library(reshape2)library(mixOmics) #PCA
Read counts
The matrix of raw counts looks like that.
Show the code
#cat("EXP = ", params$my.interest)### Get Raw Count data in edgeR object# At gene level#inputFile <- "count_allSamples_BA.csv"inputFile <-list.files("../data/counts") |>str_subset(params$my.interest)#cat("File raw = ",inputFile,"\n")rawdata <-fread(file.path("../data/counts",inputFile))# rename first column gene ID to be homogenous between assayscolnames(rawdata)[1] <-"Gene_ID"nameEXP <- params$my.interest# genecount: count matrixgeneCount <- rawdata[,-c(1)] |>as.matrix()row.names(geneCount) <- rawdata %>% dplyr::select(1) %>%unlist() %>%as.character()#row.names(geneCount) %>% head()#print(dim(geneCount))head(geneCount) %>%kbl() %>%kable_styling() %>%scroll_box(width ="100%", height ="200px")
ER_G_R1
ER_G_R2
ER_G_R3
ER_R_R1
ER_R_R2
ER_R_R3
gene-EUBREC_RS00010
2954
5651
6885
4663
4618
7345
gene-EUBREC_RS00015
1247
2482
3366
2390
2453
3605
gene-EUBREC_RS00020
147
305
378
325
374
445
gene-EUBREC_RS00025
1115
2183
2871
1876
2041
3159
gene-EUBREC_RS00030
2570
4589
5907
4570
4973
7323
gene-EUBREC_RS00035
3380
6119
8719
7076
7463
10522
This matrix contains raw counts from ER experiment with 3240 genes and 6 samples.
# GFF annotation filefilegff <-case_when( nameEXP =='BA'~"../data/GFF/NC_008618_bifido_adolescentis_spikes_cazymes_dbcan.gff3", nameEXP =='BU'~"../data/GFF/CP102263_1_Bacteroides_uniformis_spikes_cazymes_dbcan_pulpred_cc.gff3", nameEXP =='BC'~"../data/GFF/NZ_AP012325_bifido_catenulatum_spikes_cazymes_dbcan.gff3", nameEXP =='ER'~"../data/GFF/NC_012781_eubacterium_rectale_spikes_cazymes_dbcan.gff3",)# use gff produced by DBCANgff <-readGFF(filegff)df_annot <- gff %>%as_tibble() %>%unnest_longer(Parent) # remove comulmn with all NAdf_annot <- df_annot %>%select_if(~sum(!is.na(.)) >0) # Remark: duplicated gene annotation# which(duplicated(df_annot[,"Parent"]))# keeep the first line when multiple annotationdf_annot <- df_annot%>%group_by(seqid, source, type, ID, Parent) %>%filter(row_number() <=1)#glimpse(df_annot)rawdata_annot <-left_join(rawdata[, 1], df_annot, by=c("Gene_ID"="Parent"), keep=FALSE) %>%as.data.frame()#glimpse(rawdata_annot)
In this ER experiment, the effect of each treatment (G, R) on gene expression will be explore. The number of biological repetitions is 3, 3 for G, R treatment, respectively.
Genes with very low counts across all libraries may be filtered.
Show the code
# Filter gene with low count, at least number of biological replicates with cpm > valfilt #summary(libsize)#valfilt <- round(10*1e06 / min(libsize), 1)valfilt <-1nbrepbio <-min(table(colData(se)$group))keep <-rowSums(edgeR::cpm(se) > valfilt) >= nbrepbio#summary(keep)se <- se[keep,]#dim(se)
The filtering on genes is based on count per million (cpm) greater than 1 in at least 3 samples corresponding to the minimum number of biological replicates. We kept 3082 expressed genes for further analyses from 6 samples.
Normalisation
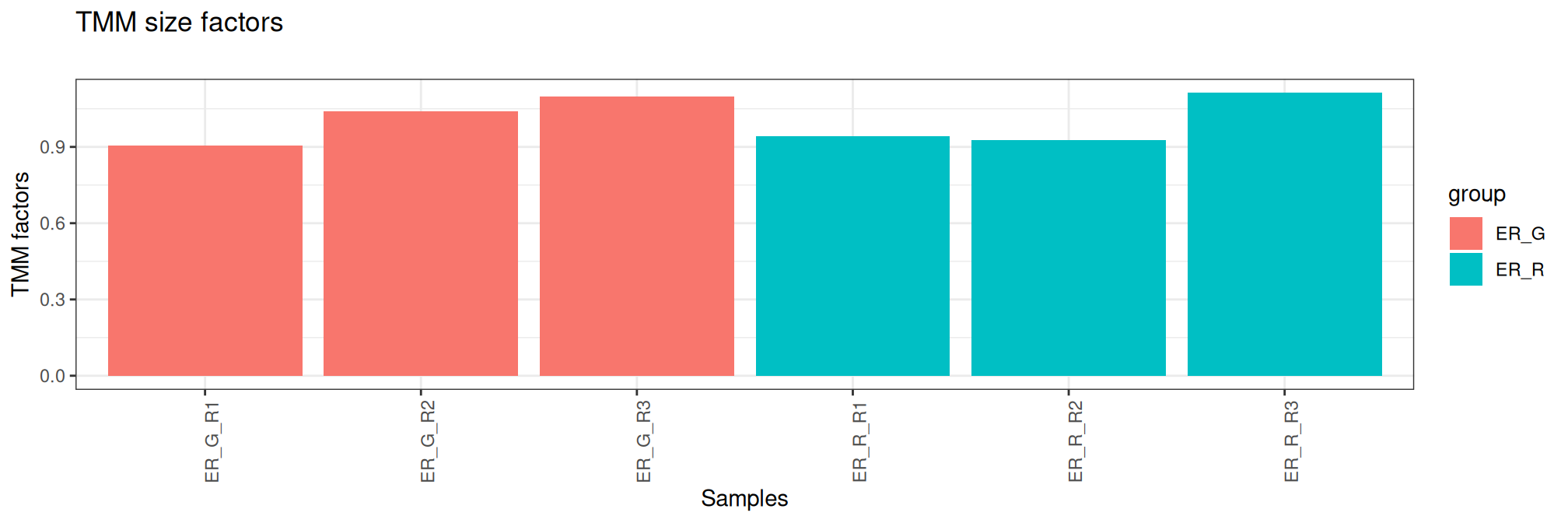
A normalization factor is calculated to take into account the different sizes of the sequencing banks (i.e. the total read count) and the distribution of reads per sample on sequencing run, as discussed [1]. Normalization by trimmed mean of M values (TMM) [2] is performed by using the calcNormFactors function from edgeR R package. It calculates a set of normalization factors, one for each sample, to eliminate composition biases between libraries.
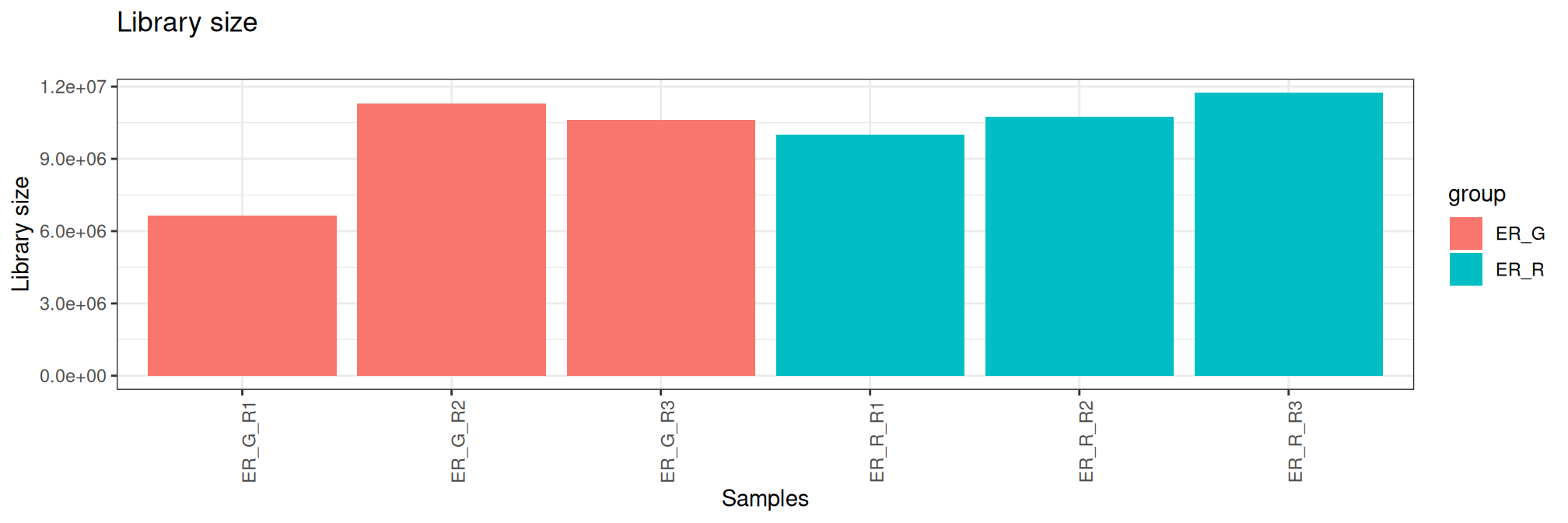
Here, the table contains library size and normalisation factors using TMM method ordered by library size. These graphs are another way of verifying the quality control carried out during the sequencing and bioinformatics analysis steps.
group lib.size norm.factors Bacteria Treatment Replicate sample
ER_G_R1 ER_G 6659374 0.9042937 ER G R1 ER_G_R1
ER_R_R1 ER_R 10014864 0.9417411 ER R R1 ER_R_R1
ER_G_R3 ER_G 10632182 1.0974065 ER G R3 ER_G_R3
ER_R_R2 ER_R 10756755 0.9262899 ER R R2 ER_R_R2
ER_G_R2 ER_G 11293072 1.0385752 ER G R2 ER_G_R2
ER_R_R3 ER_R 11735721 1.1122607 ER R R3 ER_R_R3
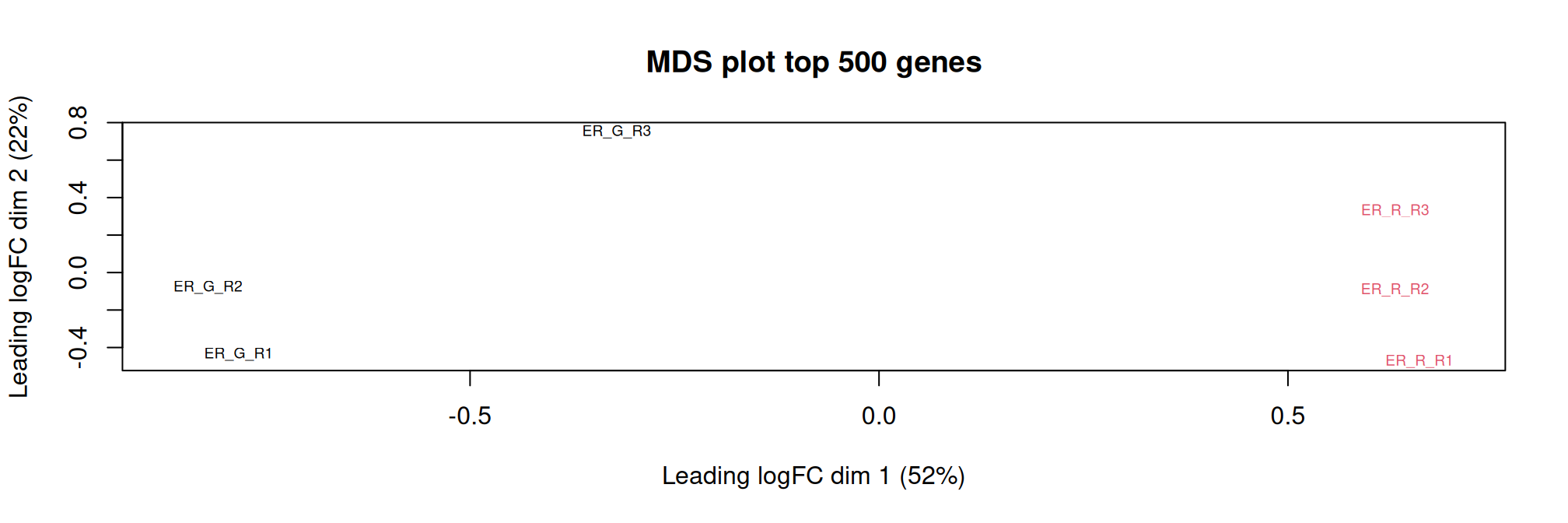
Multidimensional scaling (MDS) plot shows the relationships between the samples. The top (500 genes) are used to calculate the distance between expression profiles of samples. The distance approximate the log2 fold change between the samples.
As expected, samples are grouped by treatment.
Show the code
limma::plotMDS(dge, col =as.numeric(dge$samples$group), labels = dge$samples$sample, cex=0.6, top =500, main ="MDS plot top 500 genes")
1. Dillies M-A, Rau A, Aubert J, Hennequet-Antier C, Jeanmougin M, Servant N, et al. A comprehensive evaluation of normalization methods for illumina high-throughput RNA sequencing data analysis. Brief Bioinform. 2012;14:671–83.
2. Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biology. 2010;11:R25. doi:10.1186/gb-2010-11-3-r25.
Reuse
This document will not be accessible without prior agreement of the partners
A work by Migale Bioinformatics Facility
Université Paris-Saclay, INRAE, MaIAGE, 78350, Jouy-en-Josas, France
Université Paris-Saclay, INRAE, BioinfOmics, MIGALE bioinformatics facility, 78350, Jouy-en-Josas, France
Source Code
---params: my.interest: "BU"title: "RESTORBIOME2: Exploration and normalisation"subtitle: "Experiment with `r params$my.interest`"author: - name: Olivier Rué orcid: 0000-0003-1689-0557 email: olivier.rue@inrae.fr affiliations: - name: Migale bioinformatics facility adress: Domaine de Vilvert city: Jouy-en-Josas state: France - name: Christelle Hennequet-Antier orcid: 0000-0001-5836-2803 email: christelle.hennequet-antier@inrae.fr affiliations: - name: Migale bioinformatics facility adress: Domaine de Vilvert city: Jouy-en-Josas state: Francedate: "2023-05-02"date-modified: today#bibliography: ../../../resources/biblio.bib # don't change#csl: ../../../resources/biomed-central.csl # don't change# # Do not modify this section without taking precautions# # Don't remove the commented line, it is usefull for building the site with this new project!license: "This document will not be accessible without prior agreement of the partners"format: html: embed-resources: false toc: true toc-location: right page-layout: article code-overflow: wrap code-fold: true code-tools: true code-summary: "Show the code"---```{r}#| label = "setup",#| include = FALSEknitr::opts_chunk$set(echo =TRUE, cache =FALSE, message =FALSE, warning =FALSE, fig.height =3.5, fig.width =10.5)``````{r}#| label = "setuplib"#Use packages and functionslibrary(tidyverse)library(data.table)library(SummarizedExperiment) # manipulation RNASeq assayslibrary(rtracklayer) ## for annotation#library("DEFormats")library(edgeR)library(kableExtra)library(dplyr)library(purrr)library(stringr)library(ggplot2)library(reshape2)library(mixOmics) #PCA```# Read countsThe matrix of raw counts looks like that.```{r}#| label = "LoadRawdata"#cat("EXP = ", params$my.interest)### Get Raw Count data in edgeR object# At gene level#inputFile <- "count_allSamples_BA.csv"inputFile <-list.files("../data/counts") |>str_subset(params$my.interest)#cat("File raw = ",inputFile,"\n")rawdata <-fread(file.path("../data/counts",inputFile))# rename first column gene ID to be homogenous between assayscolnames(rawdata)[1] <-"Gene_ID"nameEXP <- params$my.interest# genecount: count matrixgeneCount <- rawdata[,-c(1)] |>as.matrix()row.names(geneCount) <- rawdata %>% dplyr::select(1) %>%unlist() %>%as.character()#row.names(geneCount) %>% head()#print(dim(geneCount))head(geneCount) %>%kbl() %>%kable_styling() %>%scroll_box(width ="100%", height ="200px")```This matrix contains raw counts from `r params$my.interest` experiment with `r nrow(geneCount)` genes and `r ncol(geneCount)` samples.This is the experimental design.```{r}#| label = "Design"# create Design from sample's names in filesampleInfo.all <-colnames(rawdata)[-c(1)] %>% stringr::str_split(., "[_]", simplify =FALSE) %>%transpose() %>% purrr::simplify_all() names(sampleInfo.all) <-c("Bacteria","Treatment","Replicate")#sampleInfo.all$sample <- paste(sampleInfo.all$Treatment, sampleInfo.all$Rep, sep="_")sampleInfo.all$sample <-colnames(rawdata)[-c(1)]Design <-as.data.frame(sampleInfo.all)Design <- Design %>%mutate( group =paste(Bacteria, Treatment, sep="_"))Design %>%kbl() %>%kable_styling() %>%scroll_box(width ="100%", height ="200px")# Design_file <- paste("../output/Design", nameEXP, ".csv", sep="")# write.table(Design, Design_file, row.names=FALSE, sep=",")```We used gene annotation produced using **DBCAN** {{< iconify mdi tools >}}.```{r}#| label = "GeneAnnotation"# GFF annotation filefilegff <-case_when( nameEXP =='BA'~"../data/GFF/NC_008618_bifido_adolescentis_spikes_cazymes_dbcan.gff3", nameEXP =='BU'~"../data/GFF/CP102263_1_Bacteroides_uniformis_spikes_cazymes_dbcan_pulpred_cc.gff3", nameEXP =='BC'~"../data/GFF/NZ_AP012325_bifido_catenulatum_spikes_cazymes_dbcan.gff3", nameEXP =='ER'~"../data/GFF/NC_012781_eubacterium_rectale_spikes_cazymes_dbcan.gff3",)# use gff produced by DBCANgff <-readGFF(filegff)df_annot <- gff %>%as_tibble() %>%unnest_longer(Parent) # remove comulmn with all NAdf_annot <- df_annot %>%select_if(~sum(!is.na(.)) >0) # Remark: duplicated gene annotation# which(duplicated(df_annot[,"Parent"]))# keeep the first line when multiple annotationdf_annot <- df_annot%>%group_by(seqid, source, type, ID, Parent) %>%filter(row_number() <=1)#glimpse(df_annot)rawdata_annot <-left_join(rawdata[, 1], df_annot, by=c("Gene_ID"="Parent"), keep=FALSE) %>%as.data.frame()#glimpse(rawdata_annot)```In this `r params$my.interest` experiment, the effect of each treatment (`r levels(as.factor(Design$Treatment))`) on gene expression will be explore. The number of biological repetitions is `r table(Design$Bacteria, Design$Treatment)` for `r levels(as.factor(Design$Treatment))` treatment, respectively.```{r}#| label = "SE"# create a SummarizedExperiment objectse <-SummarizedExperiment(assays=list(counts=geneCount),rowData=rawdata_annot, colData=Design)#print(se)#counts matrix = assay(se)#size of library#libsize <- assay(se) %>% colSums()#exemple to remove sample BT_G_Rep1#se <- se[, se$sample !="BT_G_Rep1"]#colData(se)#dim(se)#print(se)#counts matrix = assay(se)libsize <-assay(se) %>%colSums()libsize %>%kbl(col.names =c("libsize")) %>%kable_styling(full_width =FALSE)```<!-- Samples with too low depth sequencing (less than $10^7$) were removed. `r names(libsize)[which(libsize <= 10**7)]` `r libsize[which(libsize <= 10**7)]` --><!-- ```{r} --><!-- #| label = "SEremove" --><!-- #remove sample with too low depth sequencing --><!-- if (sum(libsize <= 10**7) > 0){ --><!-- se <- se[, se$sample != names(libsize)[which(libsize <= 10**7)]] --><!-- libsize <- assay(se) %>% colSums() --><!-- } --><!-- ``` -->Genes with very low counts across all libraries may be filtered. ```{r}#| label = "FiltNorm_se"# Filter gene with low count, at least number of biological replicates with cpm > valfilt #summary(libsize)#valfilt <- round(10*1e06 / min(libsize), 1)valfilt <-1nbrepbio <-min(table(colData(se)$group))keep <-rowSums(edgeR::cpm(se) > valfilt) >= nbrepbio#summary(keep)se <- se[keep,]#dim(se)```<!-- Here the cutoff of `r valfilt` for the CPM has been chosen because it is roughly equal to 10/L where L is the minimum library size in millions, for `r nbrepbio` samples corresponding to the minimum number of biological replicates. We kept `r nrow (se)` expressed genes for further analyses. -->The filtering on genes is based on count per million (cpm) greater than 1 in at least `r nbrepbio` samples corresponding to the minimum number of biological replicates. We kept `r nrow (se)` expressed genes for further analyses from `r ncol(se)` samples.# NormalisationA normalization factor is calculated to take into account the different sizes of the sequencing banks (i.e. the total read count) and the distribution of reads per sample on sequencing run, as discussed [@Dillies2012-ln]. Normalization by trimmed mean of M values (TMM) [@Robinson2010] is performed by using the `calcNormFactors` function from **edgeR** {{< iconify mdi tools >}} R package. It calculates a set of normalization factors, one for each sample, to eliminate composition biases between libraries. Here, the table contains library size and normalisation factors using TMM method ordered by library size. These graphs are another way of verifying the quality control carried out during the sequencing and bioinformatics analysis steps.```{r}#| label = "TMM"dge <-calcNormFactors(se)#cat("Ordered library size and normalisation factors TMM's method \n")# dge$samplesdge$samples[order(dge$samples$lib.size), ]# barplotp<-ggplot(dge$samples, aes(x=dge$samples$sample, y=dge$samples$norm.factors, fill=dge$samples$group)) +geom_col() +xlab("Samples") +ylab("TMM factors") +labs(fill ="group") +theme_bw() +theme(axis.text.x =element_text(angle=90)) +ggtitle("TMM size factors \n")p ``````{r}#| label = "SizeLibrary"# Library size with groupp<-ggplot(dge$samples, aes(x=dge$samples$sample, y=dge$samples$lib.size, fill=dge$samples$group)) +geom_col() +xlab("Samples") +ylab("Library size") +labs(fill ="group") +theme_bw() +theme(axis.text.x =element_text(angle=90)) +ggtitle("Library size \n")p # bp <- ggplot(dge$samples, aes(x=dge$samples$group, y=dge$samples$lib.size, fill=dge$samples$group)) + geom_boxplot() + # geom_jitter(position=position_jitter(0.1)) + theme_bw() + # theme(axis.text.x = element_text(angle=90)) +# xlab("group") + ylab("Library size") + labs(fill = "group")# bp``````{r}#| label = "Normalizeddata",#| include = FALSE# pseudo counts with log2 transformationpseudo_counts <-log2(dge$counts +1)colnames(pseudo_counts) <- dge$samples$sampledf_counts <-melt(pseudo_counts, id =rownames(pseudo_counts))names(df_counts)[1:2] <-c ("id", "sample")df_counts <-inner_join(df_counts, dge$samples[, c("sample", "group")], by=c("sample"="sample"))# pseudo normalized counts with TMM edgeR's method with log2 transformationpseudo_TMM <-log2(edgeR::cpm(dge) +1)colnames(pseudo_TMM) <- dge$samples$sampledf_TMM <-melt(pseudo_TMM, id =rownames(pseudo_TMM))names(df_TMM)[1:2] <-c ("id", "sample")df_TMM <-inner_join(df_TMM, dge$samples[, c("sample", "group")], by=c("sample"="sample"))# boxplot of pseudo countsbp <-ggplot(data=df_counts, aes(x=df_counts$sample, y=df_counts$value, fill=df_counts$group)) +geom_boxplot() +theme_bw() +ggtitle("Boxplots of pseudo counts \n") +xlab("Samples") +ylab("Library size") +labs(fill ="group") +theme(axis.text.x =element_text(angle=90)) bp# boxplot of normalized pseudo countsbp <-ggplot(data=df_TMM, aes(x=df_TMM$sample, y=df_TMM$value, fill=df_TMM$group))bp <- bp +geom_boxplot() +theme_bw()bp <- bp +ggtitle("Boxplots of normalized pseudo counts \n")bp <- bp +xlab("Samples") +ylab("Library size") +labs(fill ="group") +theme(axis.text.x =element_text(angle=90)) bp```# Multidimensional scaling plot```{r}#| label = "ACP",#| include = FALSE# with normalized countsresPCA <-pca(t(pseudo_TMM), ncomp =5)print(resPCA)plot(resPCA)plotIndiv(resPCA, group = dge$samples$group, legend =TRUE)#plotVar(resPCA, rad.in = 0.8, cex = 0.5)```Multidimensional scaling (MDS) plot shows the relationships between the samples. The top (500 genes) are used to calculate the distance between expression profiles of samples. The distance approximate the log2 fold change between the samples.As expected, samples are grouped by treatment. ```{r}#| label = "MDSplot"limma::plotMDS(dge, col =as.numeric(dge$samples$group), labels = dge$samples$sample, cex=0.6, top =500, main ="MDS plot top 500 genes")``````{r}#| label = "saveResults",#| echo = FALSEsaveRDS(se, file =paste("../output/Restorbiome_se_", nameEXP, ".rds", sep=""))saveRDS(dge, file =paste("../output/Restorbiome_dge_", nameEXP, ".rds", sep=""))```# Reproducibility token```{r}#| label = "sessionInfo"sessioninfo::session_info(pkgs ="attached")```# References